
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
POLYMERIZATION OF N,N-DIPHENYLAKRYLAMID INITIATED BY DINITRILE-AZO-DIIZOPROPYL FORMIC ACID

Полимеры, получаемые на основе азотзамещённых акриламидов, обладают рядом ценных физических и химических свойств [2]. К указанным полимерам относится и поли-N,N-дифенилакриламид (поли-ДФА), представляющий собой продукт полимеризации N,N-дифенилакриламида (ДФА) (Рис.1)
Поли-ДФА обладает фоточувствительностью и рядом других ценных свойств, позволяющих использовать его при получении электрохимических полимерных покрытий [1, 7, 10], применяться в полимерных композиционных материалах [6, 9]. Представляло интерес продолжить работы по изучению полимеризации и сополимеризации ДФА [2 – 5, 8] с целью выяснения реакционной способности мономера и установления механизма процесса.
ДФА представляет собой белый кристаллический порошок с температурой плавления 85 °С, растворимый в ацетоне, толуоле, нерастворимый в воде.
Поли-ДФА – белый или желтоватый порошок, растворимый в диметилформамиде, толуоле. В воде также нерастворим. Температура размягчения – 250-280 °С.
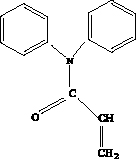
Рис. 1. Структурная формула N,N-дифенилакриламида
Материалы и методы исследования
Динитрил-азо-диизомасляной кислоты, марки «Ч» очищали двукратной перекристаллизацией из этанола. Толуол, N,N – диметилформамид, этиловый спирт, галогенопроизводные углеводородов, гексан марки «ХЧ» и «ЧДА» очищали по общепринятым методикам. Для регистрации ИК-спектров в растворе (в области 1600-1800 см-1) использовали спектрофотометр ИКС-22, растворителем служил 1,2 – дихлорэтан, не поглощающий в указанной области. Концентрация веществ составляла 0.17 моль/л. Молекулярную массу полимерных продуктов определяли в растворе толуола методом эбуллиоскопии на приборе ЭП-68. Дифференциальный термический анализ и термогравиметрический анализ проводили на дериватографе системы Паулик, Паулик, Эрдеи (Венгрия) при скорости нагрева 10о /мин. Тепловые эффекты измеряли на микрокалориметре ДАК-1-1. Для определения вязкости растворов использовали вискозиметр ВПЖ-1 с внутренним диаметром капилляра 0.34 мм. При полимеризации в растворе пробирку с рассчитанными количествами растворителя, мономера, инициатора продували азотом, очищенным от кислорода с помощью пирогаллола, плотно закрывали тщательно притёртой пробкой и нагревали до нужной температуры. Температуру поддерживали с точностью ± 0.1 °С. Обрывали полимеризацию резким охлаждением и высаживанием. При полимеризации в массе продукт растворяли в толуоле и только после этого высаживали в гексан. Для измерения тепловых эффектов реакции полимеризацию проводили в стеклянной ячейке, которая герметизировалась накидными гайками. Объём реакционной смеси составлял 4 см3. Инициатор размещался в тефлоновой капсуле, подвешенной за крышку ячейки. После достижения устойчивого экспериментального нуля микрокалориметр с ячейкой переворачивался.
Результаты исследования и их обсуждение
Попытки провести чисто термическую полимеризацию ДФА без инициатора не увенчались успехом. Термогравиметрический и дифференциальный термический анализы ДФА однозначно показали, что при его нагревании вплоть до разложения не наблюдается никаких экзотермических эффектов, которые могли бы свидетельствовать о реакции. Неполимеризуемость ДФА в отсутствие инициаторов обусловлена, вероятно, стерическими препятствиями, создающимися фенильными заместителями.
Известно, что ионная полимеризация обладает повышенной чувствительностью к малейшим примесям и, как правило, требует особых мер предосторожности при использовании инициаторов, вследствие чего её применение в промышленности весьма ограничено. Поэтому предметом нашего детального рассмотрения стала полимеризация ДФА, проводимая с помощью типичного инициатора радикального типа: динитрила–азо-диизомасляной кислоты (ДАК). Полимеризацию ДФА в присутствии ДАК проводили в массе и в растворе. В качестве растворителей использовали толуол, N,N – диметилформамид, этанол и некоторые галогенопроизводные насыщенных углеводородов: четырёххлористый углерод, хлороформ, 1,2-дихлорэтан. Контроль за выходом полимера и конверсией мономера при полимеризации в толуоле осуществлялся гравиметрически и калориметрически, по теплу, выделяемому в ходе реакции (рис. 2). Образование поли-ДФА подтверждалось исчезновением полосы поглощения 1630 см–1 (винильная группа) при сохранении полос фенильной (1630 см–1) и карбонильной (1675 см–1) групп ИК-спектра, а также результатами измерения молекулярной массы продукта (15 000 – 20 000 а.е.м.). Поскольку присутствие кислорода воздуха оказывало ярко выраженное ингибирующее влияние, то все дальнейшие кинетические исследования проводились в инертной атмосфере азота. Однако, в принципе, указанное свойство кислорода можно использовать для торможения и регулирования полимеризации ДФА в технологических процессах.
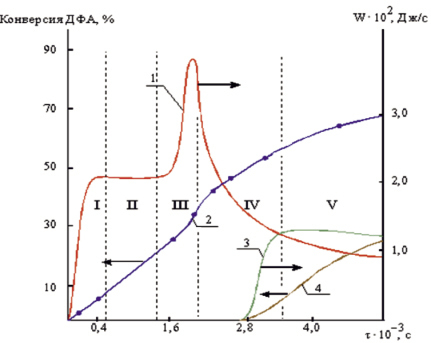
Рис. 2. Зависимости скорости (W) тепловыделения (1; 3) и конверсии мономера (2; 4) от длительности полимеризации (), определённые при [ДФА] = 0,5 моль/л, [ДАК] = 2,0 * 10-2 моль/л, t = 70 °C, в атмосфере азота (1; 2) и воздуха (3; 4)
Нестационарность начальной стадии полимеризации (участок I рис. 2) чаще всего связывают с преобладанием скорости инициирования (υи) над скоростью обрыва (υо) цепи: υи > υо. Но в данном случае ускорение обусловлено процессом растворения инициатора после его внесения в толуольный раствор мономера. Нами этот участок использовался для подтверждения значения порядка реакции по мономеру, определённого обычным методом c помощью анализа логарифмической зависимости скорости полимеризации (участок II) от концентрации мономера.
Поскольку:
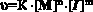 ,
,
где υ – скорость реакции, К – эффективная константа полимеризации, [М] – концентрация мономера, [I] – концентрация инициатора, n и m порядки реакции по мономеру и инициатору соответственно, то, в процессе растворения инициатора:
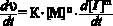
или
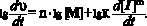
То есть, тангенс угла наклона зависимости lg dυ/ d τ) от lg[М], определённой в процессе растворения инициатора, представляет собой порядок реакции по мономеру. Для полимеризации ДФА в присутствии ДАК было установлено, что n = 1, m = 0,75. Отклонение величины m от значения , соответствующего механизму «идеальной» полимеризации, вероятно, связано со сравнительно невысокой молекулярной массой получаемого продукта, вследствие передачи цепи. Молекулярная масса поли-ДФА, полученного в растворе, не превышала 15000 – 20000 а.е.м., что соответствует степени полимеризации 70 – 90. Поэтому, как нам представляется, именно эта причина заслуживает наиболее детального рассмотрения. Так как первый порядок реакции по мономеру однозначно указывает на независимость скорости инициирования от концентрации мономера, основным фактором, вызывающим отклонение полимеризации от «идеального» течения процесса остаётся реакция передачи цепи, которая может происходить при участии любого компонента системы: инициатора, полимера, мономера, растворителя. Однако, в данном случае, в качестве инициатора использовался ДАК, константа передачи через который равна нулю. Передачей цепи через полимер, вероятно, можно пренебречь вследствие низкой концентрации последнего на начальной стадии полимеризации. Мономер также не играет существенной роли в рассматриваемом процессе, поскольку в противном случае порядок по ДФА должен быть меньше 1. Таким образом, единственно значимым передатчиком цепи в рассматриваемой системе оказывается растворитель. В таблице приведена величина общей энергии активации (Еа), которая рассчитывалась на основании уравнения Аррениуса по экспериментально определённой зависимости эффективных констант полимеризации (K) от температуры (см. таблица). Там же приведены константы инициирования (kи ) для различных температур и энергия активации инициирования (Еи).
Некоторые кинетические параметры полимеризации ДФА в толуоле, инициируемой ДАК
|
t °C |
K· 103, л3/ 4 · моль–3/ 4 · с –1 |
kи · 105, с –1 |
Еа, кДж/моль |
Еи, кДж/моль |
|
60.0 66.0 70.5 74.5 80.0 |
1.24 2.25 2.80 4.50 7.30 |
1.7 3.5 6.2 10.2 19.9 |
86 |
126 |
Полученное значение общей энергии активации (86 кДж/моль), типично для процесса радикальной полимеризации. Значения скорости и константы инициирования (υи и kи – соответственно) определяли по уравнениям:
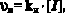
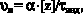
где α – коэффициент, равный количеству радикалов, обрываемых одной молекулой ингибитора (α = 1); [z] – концентрация ингибитора, τинд. – период индукции, зависящий от [z]. В качестве ингибитора использовали 1.1 – дифенил-2-пикрилгидразил (ДФПГ). Энергия активации инициирования (Еи) оказалась равной 126 кДж/моль.
Следует подчеркнуть, что приведённые результаты кинетических исследований адекватно отражают лишь начальную стадию полимеризации. Между тем, технология промышленного синтеза полимеров предполагает достижение предельных. или по крайней мере, высоких значений конверсии мономера в полимер. Из рис. 2 видно, что на определённой стадии полимеризации (участок III), несмотря на постоянство или даже замедление скорости инициирования наблюдается автоускорение процесса, наличие которого, как правило, связывают с увеличением вязкости реакционной среды в процессе полимеризации. Однако, в данном случае вязкость среды менялась незначительно.
Подобное явление, возможно связано с тем, что макромолекулы в данном случае характеризуется, прежде всего, сегментальной диффузией (а не поступательной), активных центров, уменьшение которой и приводит к эффекту автоускорения вследствие затруднения стадии обрыва цепи. Значение вязкости среды при этом может и не отличаться. Кроме того, на глубоких стадиях конверсии мономера может значительно усиливаться роль поли-ДФА в качестве передатчика цепи. При этом, радикальные центры, образующиеся в середине макромолекулы, испытывают значительно бoльшее препятствие реакции бимолекулярного обрыва, чем находящиеся на конце, что также приводит к ускорению полимеризации. Автоускорению может способствовать и формирование в растворе флуктуационной сетки, включающей растущие макрорадикалы, в результате чего у последних возникают существенные препятствия взаимной диффузии и рекомбинации. Участок IV рис. 2 соответствует прекращению автоускорения, но сохранению высокой скорости полимеризации, участок V представляет собой прекращение полимеризации вследствие исчерпания инициатора и мономера.
Немалый интерес представляет исследование влияния растворителей на процесс полимеризации ДФА. Так, имеются литературные данные о катионном механизме полимеризации некоторых азотсодержащих мономеров под действием ДАК в растворителях, представляющих собой галогенопроизводные предельных углеводородов: четырёххлористый углерод, хлороформ, 1,2-дихлорэтан. Но в данном случае нами было установлено, что при полимеризации в указанных растворителях сохраняется первый порядок реакции по мономеру, 0.7 – 0.8 по инициатору, значения Еа и энергии активации инициирования близки к величине, полученной при полимеризации в толуоле. Всё это, а также эффективное ингибирование процесса с помощью гидрохинона и ДФПГ, однозначно указывает на радикальный механизм реакции. Как видно из рис. 3 полимеризация в N,N-диметилформамиде и этаноле отличается бoльшими значениями скорости процесса, нежели в толуоле и 1.2-дихлорэтане.
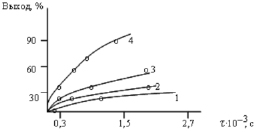
Рис. 3. Зависимость выхода поли-ДФА от продолжительности полимеризации при [M] = 0,7 моль/л, [ДАК] = 1,9•10-2 моль/л, t °C = 70 °C и различных растворителях: 1 – толуол ( = 0,36 D); 2 – 1,2-дихлорэтан ( = 1,75 D); 3 – N,N-диметилформамид ( = 3,82 D); 4 – этанол (=1,69 D)
Обнаруженное явление связано, прежде всего с увеличением константы роста цепи в полярном растворителе вследствие большей поляризации двойной связи мономера и усилении её электрофильных свойств. Косвенным подтверждением этому может служить независимость скорости инициирования от природы растворителя, установленная нами при полимеризации ДФА в присутствии ДФПГ. Но, несмотря на меньшую полярность этанола по сравнению с N,N –диметилформамидом (μ = 1.69 D и μ = 3.83 D соответственно), именно в этиловом спирте скорость процесса оказалась наибольшей. Отчасти, это можно объяснить нерастворимостью в спирте полимера, в результате чего реакция становится гетерофазной. Кроме того, этанол, являясь протогенным растворителем, способен образовывать с мономером и радикалом роста так называемый Н-комплекс ещё более усиливающий электрофильные свойства мономера и радикала, что в конечном итоге также приводит к увеличению скорости реакции.
Полимеризацию в массе исследовали гравиметрически. Пожалуй, главное, что отличает данный способ полимеризации ДФА от других – это значительные скорости, которые приводят к максимальной конверсии мономера (до 90 %) уже в течение первых пяти, десяти минут Столь быстрое протекание реакции обусловлено максимально высокой концентрацией мономера (4.5 моль/л) и высокой температурой (процесс невозможен при t °C < tпл. °C). Кроме того, полимеризация ДФА в массе практически сразу становится гетерофазной, что также приводит к ускорению процесса.
В целом, результаты исследований ДФА свидетельствуют о возможности разработки достаточно простых технологий получения фоточувствительных полимеров на его основе.
Библиографическая ссылка
Шепель О.М. ПОЛИМЕРИЗАЦИЯ N,N-ДИФЕНИЛАКРИЛАМИДА, ИНИЦИИРОВАННАЯ ДИНИТРИЛОМ-АЗО-ДИИЗОМАСЛЯНОЙ КИСЛОТЫ // Международный журнал прикладных и фундаментальных исследований. 2017. № 6-1. С. 74-78;URL: https://applied-research.ru/en/article/view?id=11625 (дата обращения: 01.07.2026).