
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
THE LONG ARM OF Y CHROMOSOME TRIPLICATION (YQ11.223Q11.23) IN A BOY WITH SPEECH DEVELOPMENT DELAY AND AUTISM.

Долгое время считалось, что структурные перестройки хромосомы Y не могут оказывать клинического эффекта в виде умственной отсталости и задержки развития, поскольку гены, вовлечённые в перестройки хромосомы Y, в основном связаны с поддержанием репродуктивных функций у мужчин. Однако с момента внедрения в диагностику методов молекулярного кариотипирования (arrayCGH, SNParray) появляется всё больше данных за то, что микродупликации и делеции хромосомы Y могут являться причиной умственной отсталости, аутизма, нарушения поведения и аномалий развития [3–5, 8, 9, 12, 17]. Стандартным кариотипированием, как правило, невозможно выявить микроструктурные перестройки хромосомы Y в силу разрешающей способности метода. В представленном нами случае перестройка была обнаружена цитогенетическим методом благодаря CBG-окрашиванию, тогда как GTG-окраска была неэффективна. Влияние структурных изменений хромосомы Y на фенотип остаются дискуссионным вопросом по той причине, что перестройки хромосомы Y редко встречаются в изолированном виде, без других изменений генома, которые также могут оказывать свое влияние на клинические проявления [5, 7, 10, 21]. Мы приводим описание мальчика с подобными геномными изменениями.
Материалы и методы исследования
В работе обследовался ребёнок мужского пола 4,5 лет. Цитогенетические исследования 72-часовой культуры лимфоцитов периферической крови с применением дифференциальной окраски хромосом по длине (GTG- и CBG-окрашивание) проводились по стандартным методикам [2]. Дополнительно ребенку проводилось молекулярное кариотипирование с использованием SNP/олигонуклеотидной микроматрицы Affymetrix Cytoscan HD, содержащей более 2,5 млн проб, с последующим биоинформатическим анализом [16].
Результаты исследования и их обсуждение
Поступивший на обследование мальчик 4,5 лет – ребёнок от 2-й беременности, протекавшей на фоне угрозы прерывания. Роды протекали срочно, стремительно. Масса тела пробанда при рождении составляла 3380 г., длина тела – 53 см. Оценка по шкале Апгар – 8/9 баллов. Голову стал держать с 1 мес., сидеть – с 7 мес., ходить – с 14 мес. Походка была неуверенная, шаткая. Во время рождения ребёнка возраст обоих родителей составлял 36 лет. Старший сибс (15 лет) здоров.
В результате обследования у ребенка были отмечены следующие клинические признаки: задержка психоречевого развития, аутистические черты в поведении, микроцефалия (окружность головы 49 см). Наблюдались такие клинические изменения, как короткая шея, S-образный грудо-поясничный сколиоз, узкая грудная клетка с килевидной деформацией, деформация нижних конечностей, плоско-вальгусные стопы, гепатоспленомегалия. Из микроаномалий развития выявлялись: краниостеноз, эпикант, рост волос на лбу в виде «мыса вдовы». При МРТ головного мозга патологии не выявлено. У ребенка отмечался аллергический ринит.
Кариотипирование по месту жительства патологии не выявило. Кариотип – 46,ХУ. Повторное цитогенетическое исследование в НИКИ педиатрии им. акад. Ю.Е. Вельтищева с использованием GTG и CBG окрашивания позволило обнаружить увеличение эухроматина в длинном плече хромосомы Y с одновременным уменьшением конституционного гетерохроматина (рис. 1, а, б, в). Эти изменения в хромосоме были видимы только при CBG-окраске (рис. 1, б). На рис. 1 для сравнения приведена нормальная хромосома Y (рис. 1, в).
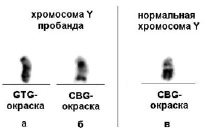
Рис. 1. Хромосома Y пробанда (а, б) и нормальная хромосома Y (в) – приведена для сравнения.
а) GTG-окраска дает гомогенное окрашивание хромосомы Y, б) CBG-окраска позволяет отобразить эухроматиновый (светлый) и гетерохроматиновый (темный) участки хромосомы. При сравнении CBG-окрашивания хромосомы пробанда и нормальной хромосомы Y (б) заметно значительное увеличение эухроматина в длинном плече и уменьшение гетерохроматинового участка в хромосоме Y пробанда
Увеличение эухроматина было предположительно расценено как дупликация. Кариотип ребенка после цитогенетического исследования: 46,Х,dup(Y)(q11.2?1q11.2?3)qh-. Для уточнения хромосомной аномалии проводилось молекулярное кариотипирование (SNParray), в результате которого увеличение эухроматина хромосомы Y было определено как трипликация Yq11.223q11.23 (геномная локализация: 24985261-28451874; размер 3.466.613 пн) (рис. 2), затронувшая 112 генов, десять из которых индексированы в OMIM: TTTY17A, TTTY4, TTTY3, BPY2, DAZ1, DAZ3, DAZ2, CDY1, CSPG4LY, GOLGA2P2Y. Перечисленные гены экспрессируются в клетках яичек и связаны с мужской фертильностью или являются псевдогенами (нефункциональными генами) (CSPG4LY, GOLGA2P2Y).
Помимо этого, были обнаружены изменения в 2 аутосомных генах: делеция в гене KYNU, локализованном в участке длинного плеча хромосомы 2 (2q22.2), размером 1782 пн, затронувшая девятый (десятый, в зависимости от изоформы) экзон этого гена, данный ген вовлечён в две геномные сети [KEGG ID: hsa00380 (tryptophan metabolism), hsa01100 (metabolic pathways)]; и делеция в гене ETS1, локализованном в участке длинного плеча хромосомы 11 (11q24.3), размером 3488 пн, затронувшая с третьего по четвертый (с седьмого по восьмой, с девятого по десятый, в зависимости от изоформы) экзоны гена ETS1. Данный ген вовлечён в пять геномных сетей [KEGG ID: hsa04014 (ras signaling pathway), hsa04320 (dorso-ventral axis formation), hsa05166 (HTLV-I infection), hsa05200 (pathways in cancer), hsa05211 (renal cell carcinoma)].

Рис. 2. Графическое отображение участка трипликации хромосомы Y после проведения молекулярного кариотипирования
Дупликации и делеции длинного плеча хромосомы Y, в участке Yq11.223q11.23, вовлечённом в трипликацию у нашего ребёнка, являются рекуррентными, неоднократно наблюдались в нашей практике и были описаны в литературе [4, 7, 9, 11]. В частности, такие перестройки были отмечены у детей с задержкой интеллектуального развития и аутизмом. Трипликации этого участка, как в нашем случае, встречаются редко. Анализ приведённых генетических изменений, обнаруженных у нашего ребёнка, указывает на то, что перечисленные 10 генов хромосомы Y, расположенные в участке трипликации и индексированные в OMIM, с малой долей вероятности могли бы быть ответственны за фенотипические проявления, поскольку связаны со сперматогенезом. Мутации в гене KYNU ассоциированы с аутосомно-рецессивным заболеванием гидроксикинуренинурией (hydroxykynureninuria [OMIM:236800]), известным как синдром Кнаппа – Комровера [13, 19, 20]. Синдром характеризуется задержкой психомоторного и речевого развития, вегетативной дистонией, церебеллярной атаксией, эпилепсией, повышенным содержанием в моче кинуренина, 3-гидроксикинуренина и ксантуреновой кислоты. Однако, учитывая наличие у ребёнка мутации лишь в гетерозиготном состоянии и отсутствие характерных для синдрома биохимических изменений в моче, говорить о данном синдроме у пробанда не представляется возможным. Ген ETS1 является прото-онкогеном и фактором транскрипции, участвующим в регуляции многих генов, которые связаны с развитием стволовых клеток, клеточным старением и гибелью клетки [15]. Ген экспрессируется в основном в лимфатических узлах и в селезёнке. Мутации в этом гене связаны с онкологическими и иммунными заболеваниями. Таким образом, в данное время обнаруженные делеции в генах KYNU и ETS1 не могут быть причиной клинических особенностей ребёнка. Однако, помимо генов, индексированных в базе OMIM, в участке Yq11.223q11.23 расположено около 100 генов, одни из которых являются псевдогенами, но другие сохраняют свою кодирующую способность. Некоторые функции этих генов недостаточно изучены, и можно сделать предположение о возможной связи этих, не индексированных в OMIM, генов с задержкой развития и аутизмом. Известно, что дупликации хромосомы Y в некоторых случаях не вызывают таких клинических признаков, какие отмечены у пробанда, но связаны с нарушением репродуктивной функции и особенностями поведения [1, 6]. В литературе известны некоторые микродупликационные синдромы, при которых родители – носители дупликации фенотипически здоровы, тогда как ребёнок, имеющий такую же дупликацию – болен. Примером может служить синдром дупликации длинного плеча хромосомы 22 (22q11.2) [OMIM:608363], при котором в 70 % случаев дупликация наследуется от одного из родителей, являющегося бессимптомным носителем [22]. Размер дупликации 22q11.2 может достигать 3 млн пн. Механизмы возникновения или отсутствия симптомокомплекса при этом синдроме неясны. Возможно, дупликации хромосомы Y также связаны с такими же, неизвестными пока, механизмами. В нашем случае у ребенка имелась трипликация, и можно предположить, что трипликации могут оказывать более значительное влияние на фенотип.
Заключение
С внедрением в генетическую диагностику метода молекулярного кариотипирования выявляемость геномных нарушений по сравнению с цитогенетическим анализом увеличилась во много раз, при этом множественные изменения генома у одного индивидуума наблюдаются достаточно часто [5, 7, 9, 18]. Однако трактовка обнаруженных генетических изменений при корреляции генотип-фенотип может быть связана с определенными сложностями, особенно в случаях сочетания нескольких аномалий. Гены, индексированные в базе OMIM, составляют только часть всех функциональных генов, значение, взаимодействие и роль в развитии организма которых пока недостаточно изучены. Остается много вопросов относительно различной проявляемости клинических признаков и их полиморфизма при одинаковой генетической аномалии у разных индивидов. Учитывая данные литературы и собственные наблюдения группы детей мужского пола с задержкой развития, аутизмом и особенностями поведения, имеющих аналогичную нашему случаю перестройку хромосомы Y, мы считаем, что основные фенотипические проявления у нашего ребёнка связаны с трипликацией хромосомы Y. Последнее время в литературе появляются данные о том, что некоторые «молчащие» гены могут быть регуляторами экспрессии генов, в частности связанными с нейродегенеративными заболеваниями, умственной отсталостью и аутизмом [14]. По-видимому, дальнейшие исследования функционирования большего числа генов и их взаимодействий, помимо генов, индексированных в базе OMIM, позволят найти ответы на многие вопросы, касающиеся корреляции генотип-фенотип в подобных случаях.
Работа поддержана грантами РФФИ (проекты № 16-54-76016 ЭРА_а и № 17-04-01366 А).
Библиографическая ссылка
Колотий А.Д., Ворсанова С.Г., Юров Ю.Б., Васин К.С., Кузнецова С.Ю., Гордеева М.Л., Юров И.Ю. ТРИПЛИКАЦИЯ ДЛИННОГО ПЛЕЧА ХРОМОСОМЫ Y (YQ11.223Q11.23) У МАЛЬЧИКА С ЗАДЕРЖКОЙ ПСИХОРЕЧЕВОГО РАЗВИТИЯ И АУТИЗМОМ // Международный журнал прикладных и фундаментальных исследований. 2017. № 8-2. С. 260-264;URL: https://applied-research.ru/en/article/view?id=11796 (дата обращения: 05.07.2026).