
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
DEVELOPMENT AND VALIDATION OF METHODOLOGY FOR QUANTITATIVE DETERMINATION OF IMPURITIES IN A SUBSTANCE «LACTAPTIN, THE RECOMBINANT ANALOG OF HUMAN MILK PROTEIN»

В ИХБФМ СО РАН из молока человека был выделен и охарактеризован белок – протеолитический фрагмент каппа-казеина человека с молекулярной массой ~8,6 кДа, получен его генноинженерный аналог, который, как и природный пептид, обладает способностью индуцировать апоптоз раковых клеток человека в культуре и тормозить рост и метастазирование опухолей животных и человека.
На основе полученного рекомбинантного белка разработан новый противоопухолевый препарат «Лактаптин, концентрат для приготовления раствора для инфузий» и успешно завершены его доклинические исследования [1].
В рамках доклинических исследований показано, что разработанный препарат является безопасным, проявляет противоопухолевую и антиметастатическую активность в исследованиях на лабораторных животных и может быть рекомендован для проведения клинических исследований. Назначение препарата – лечение рака молочной железы человека.
Активная субстанция препарата представляет собой белок Лактаптин, рекомбинантный аналог пептида молока человека, продуцируемый генетически модифицированной культурой штамма Escherichia coli BL21(DE3)/pFK2, находящийся в 0,9 % растворе натрия хлорида. Внешний вид – замороженный раствор (плотная затвердевшая белого цвета масса), после размораживания – бесцветная жидкость, прозрачная или слабо опалесцирующая.
Для обеспечения безопасности и эффективности лекарственных средств на основе рекомбинантных белков необходим тщательный контроль качества очищенного белка с использованием валидированных методик. Уровень специфических и неспеци-
фических примесей в субстанции определяется источником ее получения, технологическим процессом и является важнейшей составляющей качества препарата. Содержание примесей должно быть регламентировано и не превышать требований, указанных в нормативной документации [4].
Целью данной работы являлись разработка и валидация методики количественного определения посторонних примесей в субстанции лактаптин – методом обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ-ВЭЖХ).
Материалы и методы исследования
При подготовке нормативной документации (ФСП, регламенты) на субстанцию Лактаптин была разработана методика количественного определения посторонних примесей в лекарственном средстве методом ОФ-ВЭЖХ. Разработка и валидация методики осуществлялась на 5 экспериментальных сериях субстанции «Лактаптин, рекомбинантный аналог пептидов молока человека», разработчик и производитель – ИХБФМ СО РАН.
Количественное содержание примесей в субстанции определяли на микроколоночном жидкостном хроматографе Милихром А-02 (ЗАО Эконова, Россия), имеющем градиентную систему подачи элюента и УФ-спектрофотометрический детектор. Обработка хроматографических данных проведена с помощью программы МУЛЬТИХРОМ (фирма «Амперсенд», г. Москва).
Растворы и реактивы: ацетонитрил 1 сорта (ГОСТ 11097-86), ортофосфорная кислота концентрированная, х.ч. (ГОСТ 6552-80), вода очищенная (ФС.2.2.0020.15).
Валидацию методики количественного определения посторонних примесей в субстанции лактаптин проводили в соответствии с [2, 3] по следующим характеристикам: селективность, линейность, правильность и прецизионность.
Для проверки линейности методики использовали образец субстанции, подвергнутый с целью накопления в нем примесей частичной деградации путем воздействия экстремальных условий (температура 45 °С в течение 48 ч). Экспериментальный образец, содержащий после термического разрушения 6,85 % примесей, в дальнейшем разбавляли в растворе натрия хлорида 0,9 % до концентрации примесей 1,71 % и анализировали.
Результаты исследования и их обсуждение
Для количественного определения посторонних примесей в субстанции Лактаптин нами была разработана ОФ-ВЭЖХ методика: колонка хроматографическая Protnto «SIL-120-5-C18 AQ» (ЗАО «Эконова», Россия) размером 2,0×75 мм, заполненная сорбентом типа С18 с размером частиц 5,0 мкм (Pronto SIL 120-5-C18); подвижная фаза – элюент А: 0,1 об. % ортофосфорной кислоты в воде; элюент В: 0,1 об. % ортофосфорной кислоты в ацетонитриле; скорость потока
150 мкл/мин; температура термостата колонки 35 °С; объем вводимой пробы 5 мкл; время хроматографирования 13 мин; детектор спектрофотометрический, базовая λ = 220 нм.
Таблица 1
Градиентный режим
|
Шаг |
Регенерация |
0 |
1 |
2 |
3 |
|
Объем, мкл |
800 |
0 |
400 |
2000 |
2500 |
|
Элюент В, % |
5 |
5 |
5 |
100 |
100 |
Для приготовления рабочего раствора испытуемой субстанции в микропробирку помещали 5 мкл субстанции и 120 мкл подвижной фазы, перемешивали, затем 25 мкл полученного раствора переносили в пробирку для автодозатора и хроматографировали.
Пригодность хроматографической системы подтверждена на шести последовательных анализах стандартного образца предприятия Лактаптин (СО).
На рис. 1 представлена хроматограмма определения примесей в стандартном образце предприятия субстанции Лактаптин с использованием разработанной методики.
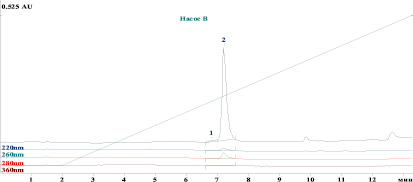
Рис. 1. Хроматограмма стандартного образца предприятия лактаптина: 1 – пик высокомолекулярных примесей, 2 – пик белка лактаптин
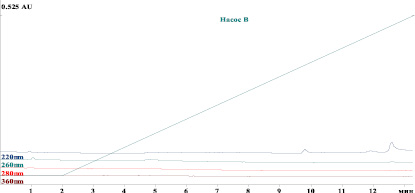
Рис. 2. Хроматограмма плацебо
На хроматограмме пики белка лактаптина и примесей хорошо разделены между собой и не мешают их определению. Среднее время удерживания белка лактаптин – 7,2 мин, примеси – 6,9 мин. При этом на хроматограммах плацебо не детектируется никаких пиков со временем удерживания, совпадающим со временем удерживания пика лактаптина (рис. 2).
Эффективность хроматографической колонки, рассчитанная по пику белка лактаптин, составляет 21000 теоретических тарелок, среднее разрешение пиков белка лактаптин и примеси 1,16, что удовлетворяет критерию пригодности хроматографической системы для анализа высокомолекулярных примесей (среднее разрешение пиков ≤ 2,0 и число теоретических тарелок ≥ 5000). Относительное стандартное отклонение (RSD) площадей пиков белка лактаптин для шести последовательных хроматограмм составило 0,87 %.
Критериями оценки качества субстанции были приняты площадь основного пика, которая должна составлять не менее 95 % от общей площади всех детектированных пиков, сумма площадей дополнительных пиков должна составлять не более 5 % от общей площади всех пиков.
Определение линейности методики проводили на пяти уровнях предполагаемых концентраций сопутствующих примесей в диапазоне концентраций от 1,71 % до 6,85 %. Модельные растворы готовили путем разбавления экспериментального образца субстанции, содержащего после термического разрушения 6,85 % примесей (табл. 2).
Таблица 2
Результаты анализа исследуемых образцов для показателя линейность
|
Номер образца |
Содержание примесей, % |
Сумма площадей пиков сопутствующих примесей, отн. ед. |
Площадь средняя, отн. ед. |
RSD |
||
|
4 |
6,85 |
0,218 |
0,213 |
0,218 |
0,216 |
1,10 |
|
5 |
4,91 |
0,146 |
0,147 |
0,148 |
0,147 |
0,54 |
|
6 |
3,42 |
0,104 |
0,102 |
0,101 |
0,102 |
1,27 |
|
7 |
2,28 |
0,065 |
0,064 |
0,063 |
0,064 |
0,78 |
|
8 |
1,71 |
0,041 |
0,041 |
0,042 |
0,041 |
1,19 |
График зависимости суммарной площади пиков примесей от концентрации имеет линейный характер и выражается уравнением y = 0,033x – 0,0129 (рис. 3).
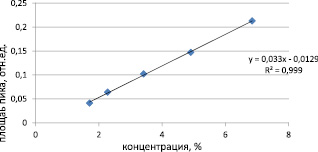
Рис. 3. Зависимость суммарной площади пиков примесей (отн. ед.) от концентрации ( %)
Достоверность линейной связи значений площади пиков примесей от концентрации в диапазоне исследуемых концентраций показана величиной коэффициента корреляции r = 0,999.
Поскольку допустимое содержание посторонних примесей в субстанции должно быть не более 5 %, следовательно, диапазон экспериментальных данных, удовлетворяющих линейной модели в интервале концентраций от 1,71 % до 6,85 %, можно рассматривать как аналитическую область. При этом наименьшая концентрация примесей в образце, которая может быть определена с использованием валидируемой методики с требуемой правильностью и прецизионностью, равна 1,71 % и соответствует критерию приемлемости (величина предела количественного определения аналитической методики ниже контролируемого предела).
Оценку правильности методики проводили по результатам анализа модельных растворов экспериментального образца субстанции, подвергнутого частичной деградации, на пяти уровнях содержания примесей. Критерием приемлемости является среднее значение % восстановления (R), которое должно находиться в пределах от 98 до 102 %. Среднее значение процента восстановления равно 100,24 % и удовлетворяет критерию приемлемости (табл. 3).
Таблица 3
Определение правильности методики
|
Заданное содержание примесей, % |
Измеренное содержание примесей, % |
R |
R ср. |
|
6,85 |
6,94 |
101,3 |
100,24 |
|
4,91 |
4,85 |
98,8 |
|
|
3,42 |
3,48 |
101,8 |
|
|
2,28 |
2,33 |
102,2 |
|
|
1,71 |
1,66 |
97,1 |
Повторяемость (сходимость) методики подтверждена по результатам анализа 6 образцов, приготовленных из одной серии субстанции Лактаптин, а также результатами анализа модельных растворов, приготовленных из экспериментального образца субстанции, подвергнутого частичной деградации, на трех уровнях концентрации примесей.
Относительное стандартное отклонение концентрации примесей на всех уровнях рассматриваемых концентраций не превышает 2,0 %, что свидетельствует об удовлетворительной сходимости результатов (табл. 4).
Таблица 4
Результаты анализа образцов для показателя повторяемость
|
Наименование образца |
Содержание примесей в образце, % Номер образца |
Среднее содержание примесей, % |
RSD |
|||||
|
1 |
2 |
3 |
4 |
5 |
6 |
|||
|
Серия субстанции |
2,08 |
2,05 |
2,06 |
1,96 |
2,05 |
1,97 |
1,97 |
0,52 |
|
Модельный раствор 1 |
6,83 |
6,94 |
6,99 |
6,85 |
7,00 |
6,99 |
6,93 |
1,09 |
|
Модельный раствор 2 |
4,82 |
4,85 |
4,86 |
4,85 |
4,86 |
4,91 |
4,86 |
0,60 |
|
Модельный раствор 3 |
3,56 |
3,47 |
3,44 |
3,44 |
3,47 |
3,56 |
3,49 |
0,61 |
Оценка внутрилабораторной прецизионности проводилась на результатах, полученных при определении повторяемости вместе с набором данных, полученных вторым химиком при выполнении анализа той же серии субстанции в другой день, на другом оборудовании, с другими реактивами. Проведена статистическая обработка результатов анализа, полученных первым химиком и вторым химиком, рассчитано относительное стандартное отклонение для примесей в субстанции, которое составило 1,9 % и удовлетворяет критерию приемлемости (не более 2 %) (табл. 5).
Таблица 5
Результаты анализа исследуемых образцов субстанции для показателя внутрилабораторная прецизионность
|
Результаты получил |
Содержание примесей в образце, % Номер образца |
|||||
|
1 |
2 |
3 |
4 |
5 |
6 |
|
|
1-ый химик |
2,08 |
2,05 |
2,06 |
1,96 |
2,05 |
1,97 |
|
2-ой химик |
1,99 |
2,01 |
2,04 |
2,00 |
2,00 |
2,03 |
|
Относительное стандартное отклонение для примесей 1,9 % |
||||||
Анализ пяти экспериментальных серий субстанции лактаптин показал, что содержание в них примесей не превышает 5,0 % (данные не представлены).
Заключение
Таким образом, разработана и валидирована ОФ-ВЭЖХ методика количественного определения посторонних примесей в субстанции «Лактаптин, рекомбинантный аналог пептидов молока человека».
Полученные результаты удовлетворяют критериям приемлемости и позволяют сделать вывод, что методика воспроизводима в условиях лаборатории и результаты, полученные с использованием данной методики, достоверны.
Методика количественного определения посторонних примесей в субстанции «Лактаптин, рекомбинантный аналог пептидов молока человека» включена в проект ФСП на субстанцию и используется для определения количественного содержания примесей в субстанции.
Библиографическая ссылка
Трошкова Г.П., Романова И.В., Кулигина Е.В., Рихтер В.А. РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИКИ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ПОСТОРОННИХ ПРИМЕСЕЙ В СУБСТАНЦИИ «ЛАКТАПТИН, РЕКОМБИНАНТНЫЙ АНАЛОГ ПЕПТИДОВ МОЛОКА ЧЕЛОВЕКА» // Международный журнал прикладных и фундаментальных исследований. 2017. № 8-2. С. 316-320;URL: https://applied-research.ru/en/article/view?id=11809 (дата обращения: 02.07.2026).