
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
SPATIAL STRUCTURE OF GENOMES OF CYANOBACTERIA

Изучение особенностей и деталей структуры нуклеотидных последовательностей является важнейшей задачей биологии в настоящее время. Исследования ведутся в двух аспектах – структурно-функциональном и эволюционном. Выявление связи между структурными компонентами и соответствующими им функциями представляет собой классическую проблему молекулярной и системной биологии, и, несмотря на обширный поток публикаций и исследований в этом направлении, она всё ещё далека от завершения. Более того, исследователи выявляют всё новые и новые структурные элементы либо новые виды и формы взаимодействий и взаимоотношений между структурными элементами биологических макромолекул, а развитие техники и инструментов исследований лишь усугубляет эту ситуацию.
Понятна важность таких исследований с точки зрения эволюционных процессов. Изучение особенностей структуры биологических макромолекул у разных организмов позволяет составить более точную картину эволюции тех или иных биологических систем – от вполне конкретных видов до экосистем и глобальных сообществ.
Кроме того, затруднения в исследованиях такого рода всегда вызывают выбор и качество того биологического материала, который берётся в рассмотрение. Дело даже не в ошибках секвенирования и/или аннотирования генетических последовательностей, неизбежных во многих случаях, а в большой сложности таких объектов, как геномы либо отдельные хромосомы. Рассматривая эти объекты, приходится анализировать набор характеристик: структуру, функцию и филогению. Эти характеристики очень сильно взаимодействуют и сильно влияют друг на друга. Причем это влияние далеко не всегда удаётся выделить в качестве отдельного и независимого фактора.
Прокариотические организмы с этой точки зрения являются более удобными объектами для исследования, чем эукариотические; геном бактерий заметно короче генома эукариот и всегда представлен одной хромосомой. Своего рода расплатой за такое удобство является заметная трудность в определении филогении бактерий, особенно для таксонов высокого уровня.
Исследование структур в генетических последовательностях также является важной задачей, осложняющейся чрезвычайно большим разнообразием структур, которые можно найти и выделить в молекулах ДНК, даже если не обращать внимания на их химические свойства. Настоящая работа посвящена изучению структур в геномах цианобактерий.
Под структурой мы будем понимать различие (либо подобие) статистических свойств отдельных формально выделяемых участков генома на уровне триплетов. Иными словами, структура, рассматриваемая в этой работе, – это взаимное расположение различных (формально выделяемых) участков генома сравнительно небольшой длины в пространстве частот триплетов, которые подсчитываются в пределах указанных участков; подробности изложены в разделе «Методы исследования».
Такой подход к изучению связи структуры геномов с их GC-составом был впервые предложен в работах Горбаня с соавторами [1, 2]. Этот же подход (с небольшой модификацией) используется и нами; один из мотивов использования метода, предложенного в указанных работах, – теория симбиогенеза [3–9]: поскольку согласно этой теории современные хлоропласты и цианобактерии имеют общего предка. Если эта теория верна, то можно надеяться найти какие-то признаки подобия структур, выделяемых как в бактериальных геномах, так и в геномах хлоропластов. Следует сказать сразу, что были обнаружены существенные различия, а не подобие. Из этого не следует, что теория происхождения хлоропластов от цианобактерий неверна; это означает, что в процессе эволюции этих двух генетических изолированных систем произошла сильная дивергенция.
Материалы и методы исследования
Введём основные понятия. Мы будем рассматривать генетическую последовательность длины L, состоящую из символов алфавита 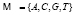 . Если последовательность содержит символы, отличающиеся от символов алфавита
. Если последовательность содержит символы, отличающиеся от символов алфавита 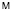 , то такие символы из последовательности удаляются, а длина последовательности уменьшается на число таких символов. Для этой последовательности символов мы будем составлять частотный словарь толщины 3. Под частотным словарем W3 толщины 3 символьной последовательности, соответствующей ДНК, будем понимать список всех триплетов v1v2v3 идущих подряд символов с указанием частот этих триплетов. Таких комбинаций может быть 64. В качестве частоты fω рассматривается отношение количества копий nω выбранного триплета к общему количеству всех триплетов N, где N – сумма всех nω:
, то такие символы из последовательности удаляются, а длина последовательности уменьшается на число таких символов. Для этой последовательности символов мы будем составлять частотный словарь толщины 3. Под частотным словарем W3 толщины 3 символьной последовательности, соответствующей ДНК, будем понимать список всех триплетов v1v2v3 идущих подряд символов с указанием частот этих триплетов. Таких комбинаций может быть 64. В качестве частоты fω рассматривается отношение количества копий nω выбранного триплета к общему количеству всех триплетов N, где N – сумма всех nω:
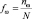 . (1)
. (1)
Любой частотный словарь W3 ставит в соответствие геному множество точек в 64-мерном метрическом пространстве. Для оценки близости двух геномов используется метрика Евклидового пространства, определяющая расстояние между двумя точками:
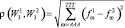 . (2)
. (2)
Для исключения линейной связи между частотами триплетов (поскольку сумма частот равна единице) один из триплетов удалялся из рассмотрения. Это позволяет уменьшить погрешность, вносимую линейной зависимостью при обработке данных статистическими методами, например при корреляционном анализе или при использовании метода главных компонент. Вообще говоря, из рассмотрения можно удалять какой угодно триплет. Тем не менее есть ряд подходов для выбора удаляемого триплета. Один из подходов в качестве удаляемого триплета предлагает выбирать триплет с максимальной частотой. Тем более если значение частоты для исключаемого триплета существенно больше (например, на порядок) значения частоты триплета, идущего за ним.
Так же часто используется подход, при котором удаляется триплет с минимальным стандартным отклонением, вычисленным по всей совокупности частотных словарей данного генома. Такой выбор обусловлен тем, что вклад этого триплета в разделение точек в пространстве минимален. В случае равенства стандартного отклонения 0 различий по этому триплету не наблюдается. Мы использовали именно этот подход. Размерность пространства уменьшается на единицу и становится 63-мерным. В рассмотренных нами геномах в большинстве случаев удалялись триплеты GCG и CGC.
Для обнаружения структуры в генетической последовательности проводилась предварительная обработка, которая ставила в соответствие данной последовательности множество точек в 63-мерном пространстве триплетов. Делалось это следующим образом: последовательность сканировалась окном длины Δ c шагом t. Для каждого положения i рамки определялся участок генетической последовательности, совпадающий с рамкой считывания, для которого вычислялся частотный словарь 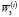 , соотносящийся с i-ой точкой 64-мерного пространства. Кроме того, с каждой точкой 64-мерного пространства связывались следующие параметры: номер центрального символа рассматриваемого участка и относительная фаза.
, соотносящийся с i-ой точкой 64-мерного пространства. Кроме того, с каждой точкой 64-мерного пространства связывались следующие параметры: номер центрального символа рассматриваемого участка и относительная фаза.
Номер центрального символа участка совпадает с номером этого символа в последовательности. Относительная фаза определяется тем, попал рассматриваемый участок в кодирующую или некодирующую область последовательности. Участок относится к кодирующим, если он целиком попадал в кодирующую область последовательности. Для некодирующего участка соответствующая ему точка помечается символом J. Если участок относится к кодирующим, для него возможны 6 вариантов маркировки: B0, B1, B2, F0, F1, F2. Для кодирующего участка, аннотированного в последовательности как считывающийся в прямом направлении, вычисляется остаток от деления на 3 разности номеров центрального символа участка и первого символа кодирующей области, к которой он относится. В соответствии с величиной остатка от деления точка помечалась символом B0, B1 или B2. Для участка, аннотированного как считывающийся в обратном направлении, вычисляется остаток от деления на 3 разности номеров последнего символа кодирующей области, к которой относится участок, и центрального символа участка. В зависимости от значения остатка от деления точка помечалась символами F0, F1 или F2. Для всех генетических последовательностей длина рамки считывания Δ = 6003, шаг t = 101.
Для полученного множества точек в программе VidaExpert [10] вычислялась и визуализировалась проекция в пространство первых трёх главных компонент из 63-мерного пространства. Для получения двумерных изображений строились проекции на плоскость первых двух главных компонент и второй и третьей главной компоненты. Группы точек, в зависимости от принадлежности участка, были обозначены разным цветом. Для некодирующих участков точки изображены на рисунках коричневым цветом, для участков, соответствующих относительным фазам B0 и F0, точки изображены темно-малиновым и светло-малиновым цветом, для участков, соответствующих относительным фазам B1 и F1, точки изображены темно-зеленым и светло-зеленым цветом, а для соответствующих относительным фазам B2 и F2 точки изображены темно-желтым и светло-желтым цветом.
Результаты исследования и их обсуждение
Гипотеза о происхождении хлоропластов от одноклеточных свободноживущих фотосинтезирующих бактерий позволяет ожидать, что структура геномов цианобактерий будет подобна аналогичной структуре хлоропластов. Мы рассмотрели структуры для геномов цианобактерий, депонированных в EMBL-банке, а именно Microcystis aeruginosa NIES-843, Nostoc sp. PCC 7107, Pleurocapsa sp. PCC 7327, Chroococcidiopsis thermalis PCC 7203, Gloeocapsa sp. PCC 7428, Anabaena cylindrica PCC 7122, Synechocystis sp. PCC 6714. Эти цианобактерии относятся к трем порядкам: Chroococcales, Nostocales, Pleurocapsales.


а) б)
Рис. 1. Структура данных Microcystis aeruginosa (а) и Nostoc sp. PCC 7107 (б) в проекции на плоскость первых двух главных компонент
На рис. 1 показан характерный вид структуры геномов цианобактерий в проекции на плоскость первых двух главных компонент. Как видно из рис. 1, структура геномов цианобактерий представляет собой своеобразные клубки из цепочек, точки в которых расположены в той же последовательности, в которой соответствующие участки расположены в геноме.

а) Проекция в плоскость первых двух главных компонент

б) Проекция в плоскость второй и третьей главных компонент
Рис. 2. Типичный вид распределения участков хлоропластных геномов наземных растений по частотам троек нуклеотидов в проекциях пространства первых трех главных компонент (приведена структура генома Lolium perenne)
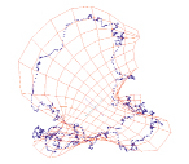
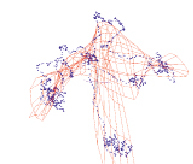
Рис. 3. Структура геномов Microcystis aeruginosa и Nostoc при шаге t, кратном трем
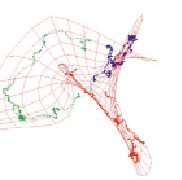
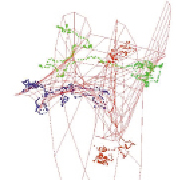
Рис. 4. Структура геномов Microcystis aeruginosa и Nostoc при шаге t, не кратном трем
Структура геномов хлоропластов наземных растений была рассмотрена в [11]. Исследование показало, что подавляющее большинство геномов хлоропластов имеет восьмикластерную структуру (рис. 2). Семь кластеров составляют трехлучевую структуру – центральное ядро из точек, соответствующих некодирующим участкам и лучи, соответствующие относительным фазам. Первый луч включает точки с маркировкой B0, F1, второй – с маркировкой B1, F0 и третий – с маркировкой B2, F2 (рис. 2, а). И есть еще выделенный кластер, который видно на рис. 2, б. Как видно из рис. 1, 2 структура геномов цианобактерий существенно отличается от структуры геномов хлоропластов. Это наблюдение может свидетельствовать о том, что расхождение от одноклеточных свободноживущих фотосинтезирующих бактерий современных цианобактерий и хлоропластов произошло очень давно и о независимом характере их развития.
Кроме того, было обнаружено, что «клубок», соответствующий структуре цианобактерий в пространстве главных компонент, состоит из одной или из трех нитей, в зависимости от величины шага t. В случае кратности шага трем клубок состоит из одной нити, в противном случае нитей три. Причем точки в одной нити следуют друг за другом последовательно, как и соответствующие им участки генома. В случае трех нитей одна нить состоит из точек, номер участков в геноме которых делится на 3 нацело, вторая нить состоит из точек, номер участков для которых делится на 3 с остатком 1, третья нить – из точек, номер участков для которых делится на 3 с остатком 2. На рис. 3 показаны участки геномов Microcystis aeruginosa и Nostoc, включающих первую тысячу точек в пространстве первых трех главных компонент для Δ = 6003 и t = 303. На рисунке видно, что структура генома представляет собой одну нить. На рис. 4 показаны участки этих же геномов для Δ = 6003 и t = 91.
Выводы
Структура геномов цианобактерий существенно отличается от структуры геномов хлоропластов. Кроме того, структура геномов цианобактерий существенно отличается и от структуры геномов иных бактерий. Семикластерная структура генома у цианобактерий отсутствует. Это также выделяет цианобактерии среди других бактерий и подчеркивает их особенности, первой среди которых выделяется способность к фотосинтезу. В частности, это наблюдение может свидетельствовать об очень древнем расхождении современных цианобактерий и хлоропластов от общего предка и о весьма сложных путях их эволюции.
Библиографическая ссылка
Сенашова М.Ю., Садовский М.Г. ПРОСТРАНСТВЕННАЯ СТРУКТУРА ГЕНОМОВ ЦИАНОБАКТЕРИЙ // Международный журнал прикладных и фундаментальных исследований. 2017. № 11-2. С. 255-259;URL: https://applied-research.ru/en/article/view?id=12009 (дата обращения: 25.06.2026).