
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
DESIGN OF MODIFIED RECOMBINANT APOLIPOPROTEIN A-I FOR TRANSFER OF NUCLEIC ACIDS

В настоящее время большую роль в области генной терапии занимают исследования по конструированию безопасных переносчиков нуклеиновых кислот на основе модульных полипептидов. Такие полипептиды состоят из различных функциональных модулей: сигналы ядерной локализации; лиганды для распознавания полипептидом определенного типа клеток; домены, конденсирующие нуклеиновые кислоты; пептиды способствующие высвобождению комплексов полипептид-НК из эндосом и другие различные модули [1, 2]. В настоящем исследовании мы предположили, что основой для такого полипептида-переносчика мог бы быть белковый компонент липопротеинов высокой плотности – аполипопротеин А-I (апоА). Многие клетки организма имеют на мембранах к апоА специфические рецепторы [3], особенно высокий уровень рецепторов был обнаружен в гепатоцитах и опухолевых клетках. Это свойство может быть использовано в качестве одной из функций полипептида на основе апоА при доставке плазмидных ДНК (пДНК) в клетки с помощью рецептор-опосредованного эндоцитоза. Второй особенностью апоА является его свойство взаимодействовать с нуклеиновыми кислотами. Так, ранее в Институте биохимии было показано, что свободный апоА, а также в комплексе с тетрагидрокортизолом связывается с эукариотической ДНК [4]. Эти предпосылки – специфическое (или слабоспецифическое) взаимодействие апоА с ДНК и рецепторный эндоцитоз апоА клетками, позволили предположить возможность использования этого белка в качестве переносчика пДНК в клетку. Мы также предположили, что можем увеличить аффинность апоА к ДНК путем введения в структуру белка фрагмента, состоящего из повторов аминокислоты лизина, имеющей положительный заряд при нейтральном рН. Известно, что фрагменты гистоновых белков, способных связываться с ДНК, богаты лизином [5]. В связи с этим целью настоящего исследования явилась модификация рекомбинантного апоА человека путем введения в его структуру полилизинового фрагмента, в также изучение способности полученного белка специфически связываться с ДНК.
Материалы и методы исследования
Для амплификации гена апоА была использована плазмида, полученная ранее в нашей лаборатории, несущая ген апоА человека [6]. Для амплификации использовали праймеры, представленные в таблице. ПЦР проводили с помощью набора реагентов с Taq ДНК-полимеразой («Евроген», Россия) на амплификаторе МС-2 («ДНК-технология», Москва). Состав реакционной смеси использовали согласно инструкции к набору. Для клонирования гена использовали модифицированный нами ранее вектор pET36b(+) «Novagen» (США) [7]. Для модификации гена в составе плазмиды pET36b(+) по сайту XhoI на 3’-конце гена апоА был встроен дуплекс, кодирующий 16 а.о., в том числе 10 а.о. лизина. Для получения дуплекса использовались олигонуклеотиды № 12 и № 13 (таблица). Олигонуклеотиды были синтезированы ЗАО «Биосан» (Россия).
Плазмиды гидролизовали эндонуклеазами рестрикции FauND I (прототип Nde I) и Sfr274 I (прототип Xho I), согласно инструкции фирмы-производителя ферментов «СибЭнзим» (Россия). Фрагменты ДНК разделяли с помощью электрофореза в агарозном геле с последующим извлечением нужного фрагмента из геля набором «Cleanup Standard» «Евроген» (Россия). Фрагменты ДНК лигировали с помощью фермента Т4 ДНК-лигазы согласно инструкции фирмы производителя «СибЭнзим» (Россия).
Трансформацию клеток E. coli плазмидными ДНК проводили с помощью электропорации по методике фирмы-производителя прибора («PeqLab, Biotechnologie GmbH», Германия). Рекомбинантные клоны E. coli отбирали на селективной среде LB (lysogeny broth), содержащей канамицин (30 мкг/мл).
Для изучения связывания белков с ДНК использовали плазмиду pTagGFP2-С размером ~ 4700 п.н. («Евроген», Россия) и амплифицированный фрагмент плазмиды pET36b(+), размером ~ 1000 п.н., содержащий ген апоА. Плазмиду нарабатывали в клетках E. coli штамм «NovaXGF» («Novagen», США) в среде LB в присутствии канамицина 30 мкг/мл. Плазмиду выделяли из клеток набором «PlasmidMidiprep» («Евроген», Россия). Фрагмент плазмиды, содержащий ген апоА, получали с помощь ПЦР с праймерами № 268F и № 13. Качество плазмиды, анализ фрагментов ДНК и продуктов ПЦР осуществляли методом электрофореза в 0,8–1,2 % агарозном геле с последующим окрашиванием ДНК бромистым этидием.
В качестве хозяйских клеток-продуцентов рекомбинантных белков использовали клетки E. coli шт. BL21(DE3). Для наработки биомассы клеток и выделения белка из отобранного клона E. coli выращивали ночную культуру в среде LB объемом 5 мл при 37 °С. На следующий день ночную культуру переносили в двухлитровую колбу с 500 мл свежей среды LB, содержащей канамицин 30 мкг/мл. Клетки выращивали при активном перемешивании и 37 °С до оптической плотности D600 = 0,8–1,2 о.е. и добавляли индуктор – изопропил-β-D-1-тиогалактопиранозид до 0,05 мМ. Далее клетки инкубировали 18 ч при 30 °С. По окончании инкубации клетки осаждали центрифугированием при 3000 об/мин в течение 20 мин, осадок замораживали и хранили до выделения белка. Клеточные лизаты и белки анализировали в 12 % полиакриламидном геле (ПААГ) по Лэммли.
Рекомбинантные белки выделяли из клеток-продуцентов E.coli с помощью аффинной хроматографии на сорбенте «Ni-NTA Superflow» («Qiagen», США) в денатурирующих условиях. Белки обессоливали методом диализа против фосфатно-солевого буфера рН 7,4–7,5 и стерилизовали фильтрованием через фильтр с размером пор 0,22 мкм.
Измерение концентрации белков и пДНК проводили спектрофотометрически в ЦКП «Спектрометрические измерения» на базе НИИ биохимии, г. Новосибирск. Измерение концентрации белков при λ = 280 нм и концентрации ДНК при λ = 260 нм проводили на спектрофотометре Evolution 300 («Thermo Scientific», США).
Результаты исследования и их обсуждение
Ранее в нашей лаборатории был получен продуцент рекомбинантного белка апоА на основе клеток E. coli [6]. Эта конструкция была использована в качестве матрицы для последовательной амплификации гена с помощью пар праймеров № 3Fа + № 6R, № 3Fb + № 6R и № 3Fс + № 6R (таблица). Это позволило ввести с 5’-конца гена фрагмент ДНК, кодирующий пептид проформы белка (Agr-His-Phe-Trp-Gln-Gln), поскольку известно, что без данного пептида уровень синтеза белка в клетках E. coli является очень низким. Праймеры № 3Fс и № 6R в своей структуре несли сайты эндонуклеаз рестрикции Nde I и Xho I, соответственно. Ампликон гена апоА был встроен в модифицированный нами вектор pET36b(+) [7], при этом на 3’-конце гена апоА, после сайта эндонуклеазы рестрикции Xho I появлялась последовательность ДНК, кодирующая 8 а.о. гистидина, что позволяло в дальнейшем выделять белок с помощью металл-хелатной аффинной хроматографии. При трансформации клеток было отобрано несколько клонов, положительных в ПЦР, несущих вставку гена апоА и способных синтезировать рекомбинантный белок при индукции клеток-продуцентов 0,05 мМ ИПТГ. Последующее выделение белка апоА из этих клонов и анализ фракций в ПААГ подтвердило правильность выбора (рис. 1, дорожки 1–3). Выход рекомбинантного апоА составил около 50 мг/л культуры клеток продуцента. Один из клонов использовался для выделения пДНК, которая в дальнейшем послужила для модификации гена апоА и введения в структуру фрагмента, кодирующего 10 а.о. лизина.
Используемые в работе олигонуклеотиды
|
Название |
Структура (5’-3’) |
|
Прямой праймер № 3Fа |
CTGGCAGCAAGATGATCCGCCGCAGAGC |
|
Прямой праймер № 3Fb |
TGCGCCATTTCTGGCAGCAAGATGATCC |
|
Прямой праймер № 3Fc |
TATCTTCATATGCGCCATTTCTGGCAGC |
|
Обратный праймер № 6R |
TACATCTCGAGCTGGGTGTTCAGCTTCTTAG |
|
№ 12 (смысловой олигонуклеотид ) |
TCGACGGCCCGGCGAAAAAGAAAAAGAAAT TAAAGAAAAAGAAAAAGC |
|
№ 13 (антисмысловой олигонуклеотид) |
TCGAGCTTTTTCTTTTTCTTTAATTTCTTTTTC TTTTTCGCCGGGCCG |
|
Прямой праймер № 268F |
ATGCGTCCGGCGTAGA |
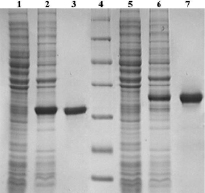
Рис. 1. Электрофореграмма анализа фракций получения рекомбинантных белков апоА и апоАpK10 из клеток E. coli в 12 % ПААГ. Дорожки: 1 и 2 – клеточные лизаты клеток-продуцентов апоА без индуктора и с индуктором соответственно; 3 – очищенный апоА; 4 – маркерные белки, бета-галактозидаза 116 кДа, бычий сывороточный альбумин 66,2 кДа, овальбумин 45,0 кДа, лактатдегидрогеназа 35 кДа, эндонуклеаза рестрикции Bsp98I 25 кДа, бета-лактоглобулин 18,4 кДа, лизоцим 14,4 кДа (Thermo Fisher Scientific, США); 5 и 6 – клеточные лизаты клеток-продуцентов апоАpK10 без индуктора и с индуктором, соответственно; 3 – очищенный апоАpK10
В полученную пДНК по сайту Xho I между геном апоА и 8 а.о. гистидина был встроен дуплекс, кодирующий 16 а.о., десять из которых представляли полилизиновый фрагмент, поделенный на две части остатком лейцина (LDGPAKKKKKLKKKKK). Дуплекс был получен путем эквимолярной гибридизации олигонуклеотидов № 12 и № 13, при этом дуплекс образовывал «липкие концы» для встройки в плазмиду, гидролизованную по Xho I сайту рестрикции. Клоны, несущие вставку полилизинового фрагмента в составе плазмиды, были отобраны методом ПЦР и проанализированы на способность синтезировать модифицированный белок апоАpK10 с помощью индукции ИПТГ и последующим анализом клеточных лизатов в ПААГ (рис. 1, дорожки 5–7). Часть положительных клонов была отобрана в музей и далее использовалась для наработки биомассы и выделения белка апоАpK10, аналогично рекомбинантному апоА.
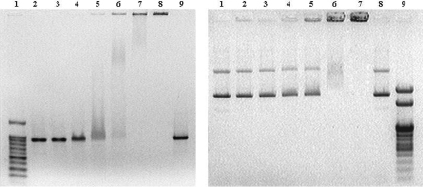
Известно, что необходимой предпосылкой для использования белка в качестве переносчика нуклеиновых кислот является его специфическое взаимодействие с ними. Одним из способов определения взаимодействия белка с ДНК является метод ретардации фрагментов ДНК в агарозном геле. Задержка ДНК в присутствии белка свидетельствует об образовании комплексов белок – ДНК, двигающихся с уменьшенной скоростью относительно свободной ДНК, либо полностью останавливающихся в геле. Для анализа мы использовали амплифицированный методом ПЦР фрагмент плазмиды размером ~1000 п.н. и плазмидную ДНК pTagGFP2-С размером ~4700 п.н. Исследовались различные соотношения по массе белок/ДНК, от 0,5:1 до 16:1. Смеси инкубировали в течение 15 минут при комнатной температуре в фосфатно-солевом буфере при комнатной температуре, затем к образцам добавляли 1/10 часть 100 % глицерина, образцы сразу же вносили в карманы геля и проводили электрофорез. Анализ взаимодействия апоАpK10 и ДНК показал, что полная задержка фрагментов ДНК и плазмиды в карманах геля происходила при избытке белка в ~16 раз (рис. 2, А, дорожка 8 и рис. 2, Б, дорожка 7).
В качестве контрольного анализа аналогичным образом было исследовано взаимодействие рекомбинантного апоА не содержащего полилизинового фрагмента. Результаты показали отсутствие изменений подвижности фрагментов ДНК в геле, даже при избытке белка по массе в 16 раз (данные не приводятся). Таким образом, задержка фрагментов ДНК в агарозном геле свидетельствует о том, что модифицированный апоАpK10, за счет введения в его структуру фрагмента из 10 а.о. лизина, приобрел способность конденсировать на себе ДНК, т.е. образовывать комплекс белок – ДНК. Из литературных данных известно, что гистоновые белки, эффективно переносящие ДНК в клетки млекопитающих, осуществляют полную задержку плазмиды в карманах геля уже при соотношении белок – ДНК равном 1:1, например Н1С гистон [8]. Возможно, что введение в структуру белка 10 а.о. лизина оказалось недостаточным для эффективного переноса ДНК в клетки.
А Б
Рис. 2. А. Электрофореграмма анализа смесей амплифицированных фрагментов плазмиды и апоАpK10. Дорожки: 1 – ДНК маркер 100–1000 и 1500 п.н. («СибЭнзим», Россия); 2 и 9 – фрагменты без инкубации с белком; 3–8 – фрагменты, инкубированные с избытком белка по массе в 0,5, 1, 2, 4, 8 и 16 раз соответственно. Б. Электрофореграмма анализа смесей плазмиды pTagGFP2-С и апоАpK10. Дорожки: 1 и 8 – контрольная плазмида, инкубированная без апоАpK10; 2–7 – образцы плазмиды, инкубированные с избытком апоАpK10 по массе в 0,5, 1, 2, 4, 8 и 16 раз соответственно; 9 – ДНК маркер 100–1000 п.н., 2 и 3 т.п.н. («СибЭнзим», Россия)
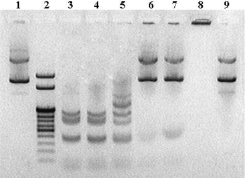
Рис. 3. Электрофореграмма анализа смесей плазмиды pTagGFP2-С и апоАpK10 в присутствии Hinf I. Дорожки: 1 и 9 – контрольная плазмида, инкубированная без апоАpK10; 2 – ДНК маркер 100–1000 п.н., 2 и 3 т.п.н. («СибЭнзим», Россия); 3 – плазмида, инкубированная с ферментом; 4–7 – образцы плазмиды, инкубированные с ферментом, в присутствии белка апоАpK10 с избытком по массе в 2, 4, 8 и 16 раз соответственно; 8 – плазмида, инкубированная с избытком апоАpK10 по массе в 16 раз, в пробу не добавляли додецилсульфат натрия
Известно, что для успешного применения белка в качестве переносчика нуклеиновых кислот в клетки млекопитающих белок должен защищать переносимый материал от нуклеаз [1, 2]. В частности, трансфекция обычно проводится в присутствии 10 % фетальной бычьей сыворотки, которая является источником нуклеаз. Чтобы смоделировать подобную ситуацию, мы решили изучить взаимодействие плазмиды pTagGFP2-С с апоАpK10 в присутствии эндонуклеазы рестрикции Hinf I. В результате полного гидролиза плазмиды ферментом должно образовываться 13 фрагментов размером 1010, 860, 782, 464, 442, 410, 396, 134, 58, 55, 52, 37 и 22 п.н. Как и в предыдущем эксперименте, белок инкубировали с плазмидой, затем в пробы добавляли по 1 ед. фермента и продолжали инкубацию проб при 37 °C в течение 30 мин. После чего, для диссоциации комплексов белок – плазмида, к пробам добавляли додецилсульфат натрия до 0,5 % по объему и вносили пробы в карманы геля. Результаты анализа представлены на рис. 3.
Анализ взаимодействия апоАpK10 с плазмидой в присутствии эндонкулеазы рестрикции Hinf I показал, что при малом избытке белка (в 2 раза) плазмида полностью гидролизовалась, аналогично плазмиде, инкубированной без белка (рис. 3, дорожки 3 и 4). При избытке белка в 8 и 16 раз плазмида сохраняла исходную форму и не подвергалась гидролизу.
Заключение
В результате выполнения работы получен модифицированный апоА-I человека, содержащий на С-конце 10 а.о. лизина. Полученный белок апоАpK10 дозозависимым образом взаимодействовал с ДНК и защищал ее от нуклеазы Hinf I, что свидетельствует о специфичности взаимодействия. Полученный белок может быть перспективным переносчиком нуклеиновых кислот в клетки организма.
Библиографическая ссылка
Котова М.В., Рябченко А.В., Трифонова Н.В., Князев Р.А., Поляков Л.М. ПОЛУЧЕНИЕ МОДИФИЦИРОВАННОГО РЕКОМБИНАНТНОГО АПОЛИПОПРОТЕИНА А-I ДЛЯ ПЕРЕНОСА НУКЛЕИНОВЫХ КИСЛОТ // Международный журнал прикладных и фундаментальных исследований. 2017. № 12-2. С. 317-321;URL: https://applied-research.ru/en/article/view?id=12042 (дата обращения: 01.07.2026).