
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
THE SEVERE CARDIAC AUTONOMIC NEUROPATHY IN PATIENT WITH DIABETES MELLITUS

Автономная кардиальная нейропатия (АКНП) представляет собой синдром крайне выраженного угнетения вегетативной регуляции синусного узла сердца (СУ), формирующийся в условиях хронической гипергликемии [1–3]. Это часто встречающееся осложнение сахарного диабета (СД), в том числе у молодых пациентов, увеличивает смертность больных с СД в среднем в 3 раза [4, 5]. Диагностика АКНП основывается на исследовании вариабельности ритма сердца (ВСР). При использовании высокочувствительной ритмокардиографии (РКГ) для анализа ВСР с помощью оценки изменчивости временных межсистолических интервалов сердца определяется крайняя выраженность угнетения автономной регуляции со стабилизацией сердечного ритма и снижением или отсутствием реагирования на стимулы [6, 7]. Считается, что в детском возрасте АКНП встречается достаточно редко, но, по мнению ряда авторов, её формирование начинается уже в подростковом возрасте [8, 9]. До настоящего времени являются единичными и весьма противоречивыми сведения о распространенности вегетативных дизрегуляций у пациентов СД 1 типа детского возраста, что связано с отсутствием клинически значимых проявлений и методологическими трудностями доклинической диагностики АКНП у детей [9, 10]. Это объясняет актуальность поиска новых методов диагностики данного осложнения. У большинства авторов синдром АКНП трактуется как автономная денервация СУ на синаптическом уровне передачи импульсов с окончаний вегетативных нервов клеткам-эффекторам, в данном случае – пейсмейкерам СУ сердца [4, 11, 12]. Гистоморфологическая основа данного состояния является недостаточно изученной [11–13].
Больная А., 16 лет, 1 октября поступила в реанимационное отделение центральной районной больницы (ЦРБ) с клиникой декомпенсации СД на фоне острой респираторно-вирусной инфекции (ОРВИ), острого ларинготрахеита, с жалобами на затруднение дыхания, одышку, головную боль, общую слабость, приступы сердцебиения, жажду, боли в животе, тошноту, повышение температуры тела до 38 °С. При осмотре ЧСС 120–140 уд/мин (периодически пароксизмы наджелудочковой тахикардии до 240 уд/мин), ЧДД 30–40 в мин. Больная была в сознании, правильно отвечала на вопросы, из лабораторных обследований – лейкоцитоз до 19,8/109/л, гликемия 8,8–15,0 ммоль/л, ацетонурия.
В связи с отсутствием эффекта от лечения на уровне ЦРБ (в/в капельно инсулин, цефабол, р-ры NaCl 0,9 %, глюкозы 5 %, KCl 5 %, сульфат магния, обильное щелочное питьё) 2 октября переведена в отделение реанимации и интенсивной терапии Челябинской областной клинической больницы (ОРиИТ), жалобы и анамнез собрать не удалось из-за ухудшения состояния и неконтактности больной. Из амбулаторной карты известно, что пациентка страдала СД в течение 6 лет (с 10-летнего возраста). Инсулинотерапия в режиме многократных инъекций (последние дозы протафана 14 единиц перед завтраком + 14 единиц в 22:00; актрапида по 8–10 единиц перед основными приёмами пищи). Диету соблюдала не всегда, компенсации СД на амбулаторном этапе не было. Гликированный гемоглобин (HbA1c) – 10,2 %. Диабетическая полинейропатия в течение трёх лет. Диабетическая непролиферативная ретинопатия и диабетическая нефропатия, стадия протеинурии – около 1 года.
При объективном обследовании: пациентка в прекоматозном состоянии, кожные покровы и видимые слизистые пониженной влажности, язык суховат, диабетический рубеоз лица, запах ацетона в выдыхаемом воздухе. ЧДД 30–32 в минуту, в лёгких дыхание везикулярное, проводится по всем лёгочным полям, хрипов нет. Тоны сердца приглушены, ритм правильный, ЧСС 113 уд/мин. АД 130/60 мм рт. ст. Живот мягкий, реагирует на пальпацию в эпигастрии, край печени выступает на 2 см из-под реберной дуги. Пастозность голеней.
Данные лабораторных методов исследования: гипергликемия до 28–35,0 ммоль/л. Выраженный гиперосмолярный синдром (натрий 156–170 ммоль/л), выраженная гипокалиемия до 2,0–2,9 ммоль/л, метаболический ацидоз (рН до 6,48; BE – 22 ммоль/л).
Данные инструментальных методов исследования
Рентгенография органов грудной клетки: снижение пневматизации лёгочной ткани в нижних отделах правого легкого, органы средостения не изменены. ЭКГ: синусовая тахикардия 130 уд/мин, поворот сердца против часовой стрелки, элементы нарушения внутрижелудочковой проводимости, выраженные изменения миокарда желудочков. ЭХОКГ: митрально-папиллярная дисфункция с регургитацией 1 степени, легочная гипертензия (СДПЖ 35 мм рт.ст.), камеры сердца не увеличены, сократительная способность миокарда сохранена.
При исследовании ВСР в покое (Ph) и пробе Ашнера – Даньини, выявлено: фиксированная тахикардия до 140 уд/мин (RR 0,438 c), крайне выраженное угнетение рефлекторной симпато-парасимпатической регуляции в синусном узле сердца со снижением общей вариабельности – σRR(SDNN) до 5 мс при норме в 50 мс, указывающее на высокий риск летального исхода [4, 9, 10]. Наблюдается «стабилизация» ритма сердца, что выглядит как прямая линия из RR-интервалов на экране монитора, отсутствие реакции на стимулы в пробе Ашнера – Даньини. В спектре регуляции преобладает «пик» непарасимпатических высокочастотных волновых колебаний в диапазоне 0,2 Гц, представляющих низкоампилитудное удлинение 3–4х RR-интервалов, связанное с эндогенной интоксикацией (рис. 1).
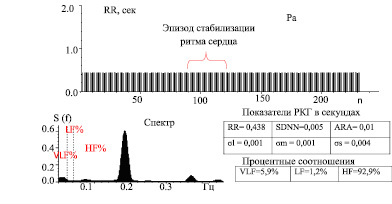
Рис. 1. Ритмокардиограмма пациентки с СД1 в пробе Ашнера (Pa) с показателями ВСР: SDNN(с) – общая вариабельность, АRA(с) – амплитуда дыхательной аритмии, среднеквадратические отклонения гуморально-метаболических – бl(с), симпатических – бm(с), парасимпатических -бs(с) волн и их процентные соотношения (VLF, LF, HF; %)
Ультразвуковое исследование органов брюшной полости: умеренная гепатомегалия, диффузные изменения печени, поджелудочной железы, почек. Люмбальная пункция – ликвор красного цвета, после центрифугирования – ксантохромный, слегка мутный, белок 1,6 г/л, эритроциты 0,09/1012/л, цитоз 28160/3, преобладают нейтрофилы. Больная проконсультирована хирургом, офтальмологом, гинекологом, данных за соответствующую патологию не обнаружено.
Проведенная терапия: внутривенно актрапид инфузоматом со скоростью 3–10 ед. в час, солевые растворы, глюкоза 5 %, гелофузин 500 мл, цитофлавин 10,0 г, берлитион 600 мг; дофамин для поддержания гемодинамики; антибиотики в/в – абактал 0,4 г два раза в сутки, роцефин – 2 г в сутки, метрогил – 0,5 г два раза в сутки.
Состояние пациентки без положительной динамики. На фоне прогрессирующей полиорганной недостаточности, метаболического ацидоза и водно-электролитных расстройств развилась 4.10 в 01:40 выраженная брадиаритмия с переходом в фибрилляцию желудочков и асистолию, после проведения реанимационных мероприятий гемодинамика восстановилась. В 4:10 вновь был эпизод гипосистолии, в/в веден атропин 1 % 1 мл и адреналин 0,5 мл 1 % раствора – гемодинамика стабилизировалась. В 9:30 развилась гипосистолия, артериальное давление не определялось, начатые реанимационные мероприятия оказались неэффективны, в 10:00, через 2 суток с момента поступления в ОРиИТ, зафиксирована биологическая смерть.
Заключительный клинический и патологоанатомический диагноз
Сахарный диабет 1 типа, тяжелое течение, декомпенсация с кетоацидотической комой: атрофия, липоматоз поджелудочной железы, жировая дистрофия печени, плазматическое пропитывание, гиалиноз артерий кишечника, желудка, почек, поджелудочной железы, селезёнки, диабетический гломерулосклероз (диффузный и узелковый гломерулонефрит), диабетическая ретинопатия, полинейропатия (по клиническим данным). Атеросклероз аорты второй стадии, 2 степени. Артериальная гипертензия 2 стадии (гипертрофия левого желудочка, толщина миокарда левого желудочка 1,5 см), отёк – набухание головного мозга (масса головного мозга 1350 г, периваскулярный, интрацелюллярный, межклеточный отёк по данным микроскопического исследования), отёк лёгких (масса легких 1160 г, диффузное заполнение альвеол эозинофильным содержимым по данным микроскопического исследования).
По протоколу патологоанатомического исследования непосредственной причиной смерти признана диабетическая кетоацидотическая кома. Причиной жизнеугрожающих, рецидивирующих нарушений ритма сердца могла явиться диагностированная прижизненно тяжёлая АКНП, для ответа на этот вопрос было проведено исследование тканей в области синусного узла методом просвечивающей электронной микроскопии (ПЭМ).
Данные гистоморфологического исследования аутопсийных препаратов тканей в области синусного узла
На светооптическом уровне выявлены признаки необратимых дистрофических и некробиотических изменений пейсмейкерных клеток синусового узла (рис. 2, А) и волокон Пуркинье (рис. 2, Б).
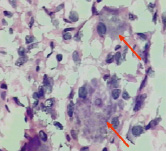
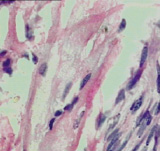
А Б
Рис. 2. А) Пейсмейкерные клетки синусового узла. Выраженные дистрофические и некробиотические изменения (стрелка); Б) Дистрофические изменения в клетках типа Пуркинье проводящей системы. Окраска гематоксилин-эозин. Ув. х 400
С помощью ПЭМ выявлены выраженные дистрофические изменения проводящей системы. Резко выражен периваскулярный, перицеллюлярный отек. Повреждения энергетического аппарата клетки в виде гиперплазии и деструкции митохондрий, накопления пигмента старения – липофусцина, кальцинаты в митохондриях связаны с основным заболеванием (рис. 3, А, Б).
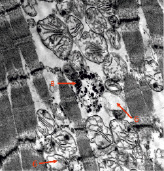

А Б
Рис. 3. А–Б. Клетки переходного типа из зоны атриовентрикулярного узла. Набухание митохондрий, разрывы крист (а). Деструкция мембран с образованием липидных капель (б). Скопления липофусцина (в). Кальцинаты митохондрий (г). КПЭМ. Ув. х 5200
Повреждения щелевых контактов вставочных дисков могут представлять собой фактор для развития нарушений проводимости (рис. 4, А, Б). Данные изменения свидетельствуют о хроническом прижизненном энергодефиците тканей в области синусного узла и проводящей системы сердца.
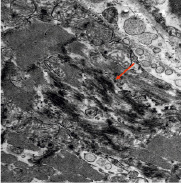
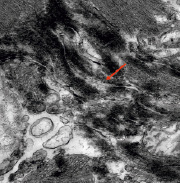
А Б
Рис. 4. А) Вставочные диски в сократительном миокарде (стрелка). Углубление интердигитаций, расширение нексусов, щелевых контактов. КПЭМ. Ув. х 4000. Б) Вставочные диски в сократительном миокарде при большем увеличении (стрелка). Десмосомальные контакты сохранены. Интерстициальный отёк. КПЭМ. Ув. х 10000
Гистоморфологические изменения, подобные описанным в данном клиническом случае, но на большем количестве аутопсийного материала, были представлены в 2011 г. в объеме комплексного исследования больных (n = 380) с профессиональными заболеваниями (ПЗ) с изучением ВСР, ЭХОКГ, а также биохимическим определением продуктов липопероксидации и процессов окислительной модификации белков [11, 12]. Корреляционный анализ выявил взаимосвязи между снижением показателей ВСР, ЭХОКГ признаками ремоделирования левого желудочка сердца и биохимическими маркерами оксидантного стресса [12]. Нарушения свободно-радикального окисления были расценены как симптомы мембранопатологических расстройств, обязательно сопровождающих процессы ремоделирования тканей сердца. Синусный узел и элементы проводящей системы сердца исследовали гистологически и электронномикроскопически после гибели 14 пациентов, имевших при жизни автономную кардионейропатию на фоне ПЗ. Полученные изменения дистрофического характера в клетках проводящей системы и синусового узла были лишены специфичности [11–13].
Таким образом, анализируя случаи гибели пациентки с СД и больных разными этиопатогенетическими формами ПЗ, можно подтвердить, что АКНП – это неспецифичный синдром, который может быть обусловлен не только функциональными, но и морфологическими изменениями в пейсмейкерах синусового узла, хотя сам термин «дизрегуляции», принятый у вегетологов, указывает только на функциональность.
Выводы
Удалось подтвердить наличие у пациентки нарушений вегетативной регуляции ритма сердца крайней степени выраженности в виде диабетической кардиальной автономной нейропатии. Обнаруженные изменения СУ и проводящей системы сердца были значительными и могли явиться субстратом для развития жизнеугрожающих нарушений ритма и смерти больной.
Библиографическая ссылка
Нуждина Е.В., Давыдова Е.В., Уточкина И.М., Миронов В.А., Тюльганова В.Л. АВТОНОМНАЯ КАРДИАЛЬНАЯ НЕЙРОПАТИЯ С ЛЕТАЛЬНЫМ ИСХОДОМ У ПАЦИЕНТКИ С САХАРНЫМ ДИАБЕТОМ 1 ТИПА // Международный журнал прикладных и фундаментальных исследований. 2018. № 5-2. С. 364-369;URL: https://applied-research.ru/en/article/view?id=12270 (дата обращения: 31.07.2026).