
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
THE REACTIONS OF 2,6-SUBSTITUTED 1,4,3,5-OXATHIADIAZIN-4,4-DIOXIDES WITH AROMATIC AND UNSATURATED REAGENTS

Памяти профессора Александра Андреевича Мичурина
Осуществление направленных синтезов сульфонилазотсодержащих соединений непосредственным взаимодействием цианосодержащих соединений и триоксида серы затруднено вследствие низкой селективности протекающих в этой системе реакций. При использовании в синтезах связанных форм SO3 (комплексов и аддуктов нитрилов с SO3) удается, во-первых, значительно повысить селективность реакций, а во-вторых, реализовать направления реагирования, не характерные для прямого взаимодействия цианосодержащих соединений и SO3 [1, 2]. Сульфонилазотсодержащие соединения ациклического строения и гетероциклические производные обладают значительной биологической активностью. Они находят применение в качестве лекарственных препаратов [3–5], сладких синтетических веществ [6, с. 85–91, с. 106], пестицидов и фунгицидов нового поколения [7–9]. Большие потенциальные возможности в синтетическом плане 2,6-дизамещенных 1,4,3,5-оксатиадиазин-4,4-диоксидов (I) и малая их изученность открывают доступ к созданию новых путей синтеза новых классов и труднодоступных циклических и ациклических сульфонилазотсодержащих соединений, обладающих полезными свойствами.
Цель исследования: изучение синтетических возможностей 2,6-дизамещенных 1,4,3,5-оксатиадиазин-4,4-диоксидов (I) с различными заместителями и возможного механизма реакций при взаимодействии их с ароматическими и непредельными соединениями.
Материалы и методы исследования
В исследовании использованы диоксиды (I), имеющие в гетероцикле один сильноакцепторный заместитель (R1 = CCl3, CBr3) и второй – менее акцепторный или донорный (R2 = 2,4-Cl2C6H3, 4-ClC6H4, C6H5, CH3). Ароматические реагенты представлены соединениями бензоидного и небензоидного типов (толуол, анизол, тиофен, N,N-диметиланилин, мезитилен). Непредельные реагенты ̶ алкены различного строения: циклического, ациклического с заместителями как небольшого объёма, так и создающими стерические затруднения (циклогексен, стирол, 3,3-диметилбутен-1), фенилацетилен.
Состав и строение полученных в настоящей работе соединений доказаны данными элементного анализа, ИК, ЯМР 1Н спектров и химическими превращениями. ИК-спектры соединений записаны на спектрофотометре Specord 80-M в метиленхлориде. Спектры ЯМР 1Н записаны на спектрометре Gemini 300 (рабочая частота 300 МГц) в ДМСО-d6, внутренний стандарт – ГМДС.
Индивидуальность полученных соединений и ход реакции контролировалась методом ТСХ на пластинках Silufol UV-254, элюент – ацетон-гексан (1:1 по объему), проявление парами йода.
Результаты исследования и их обсуждение
В настоящей работе было показано, что взаимодействие диазинов (I) с аренами, алкенами и фенилацетиленом приводит к образованию соответственно N-ациларенсульфонамидов (III), насыщенных и ненасыщенных 1,4,5-оксатиазин-4,4-диоксидов (IV, V).
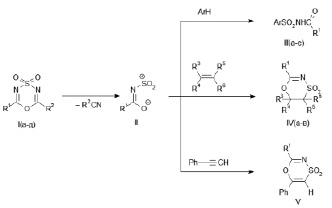
I, R1 = CCl3; R2 = CH3 (а); R1 = CCl3; R2 = 2,4-Cl2C6H3 (б); R1 = CCl3; R2 = 4-ClC6H4 (в);
R1 = CCl3; R2 = C6H5 (г); R1 = CBr3; R2 = CH3 (д);
III, R1 = CCl3; Ar = (CH)4S (а); R1 = CBr3; Ar = 4-CH3OC6H4 (б);
R1 = CCl3; Ar = 2,4,6-(CH3)3C6H2 (в); R1 = CCl3; Ar = 4-[(CH3)2N] C6H4 (г);
R1 = CCl3; Ar = 4-CH3OC6H4 (д); R1 = CCl3; Ar = 4-CH3C6H4 (е);
IV, R1 = CCl3; R3 = R6 = Н; R4R5 = -(CH2)4 (а); R1 = CCl3; R3 = R5 = R6 = Н; R4 = C6H5 (б);
R1 = CCl3; R3 = R5 = R6 = Н; R4 = (CH3)3С (в);
V, R1 = CCl3.
Рассмотрим серию опытов с использованием ароматических реагентов. При нагревании диоксидов (Iа-г) в бензоле (50 °C, 6 ч) с эквимольными количествами тиофена, N,N-диметиланилина, мезитилена происходит введение амидосульфонильного фрагмента в ароматическое кольцо с сохранением двойной связи, приводящее к образованию N-ациларен-сульфонамидов (III а, в, г). При нагревании диоксидов (I а-д) с толуолом и анизолом (110 °C, 5 часов) получены N-ациларенсульфонамиды (III б, д, е). В синтезах с толуолом и анизолом арен выступал и как реагент, и как растворитель. По истечении времени реакции растворитель отгонялся вакуумированием реакционной массы, остаток сначала нейтрализовался насыщенным водным раствором Na2CO3, затем переосаждался из нейтрального водного раствора соляной кислотой.
Выходы, температуры плавления, ИК-спектры полученных соединений (IIIа-е) приведены в табл. 1, данные элементного анализа – в табл. 2.
Таблица 1
Выходы, температуры плавления и ИК-спектры N-ациларенсульфонамидов (III а-е)
|
№ соединения |
Выход, % |
Т. пл., °С |
ИК-спектр, v, см-1 |
||
|
SO2 |
C=О |
N-H |
|||
|
III а |
98 |
154 |
1150, 1360 |
1745 |
3310 |
|
III б |
93 |
153 |
1150, 1380 |
1750 |
3390 |
|
III в |
96 |
127 |
1135, 1410 |
1750 |
3390 |
|
III г |
85 |
230 (разл.) |
1135, 1360 |
1730 |
3280 |
|
III д |
92 |
168 |
1175, 1385 |
1785 |
3390 |
|
III е |
89 |
141 |
1185,1400 |
1755 |
3390 |
Таблица 2
Данные элементного анализа N-ациларенсульфонамидов (III а-е)
|
№ соединения |
Найдено/вычислено, % |
Формула |
|
|
Эквивалент нейтрализации |
N |
||
|
III а |
306.40 306.80 |
4.43 4.54 |
C6H4Cl3NO3S2 |
|
III б |
465.51 465.70 |
3.03 3.01 |
C9H8Br3NO4S |
|
III в |
345.20 344.50 |
4.12 4.07 |
C14H12Cl3NO3S |
|
III г |
348.06 348.67 |
8.02 8.10 |
C10H11Cl3N2O3S |
|
III д |
334.31 332.50 |
4.30 4.21 |
C9H8Cl3NO4S |
|
III е |
315.80 316.50 |
4.39 4.43 |
C9H8Cl3NO3S |
Для доказательства состава и строения полученных соединений (III) кроме методов ИК-спектроскопии и данных элементного анализа проведено химическое превращение. Так, при выдерживании раствора N-трихлорацетил-2-тиофенсульфонамида (IIIа) в CH2Cl2 над увлажненным водой γ-Al2O3 (48 ч, 15 °C) из раствора выделен 2-тиофенсульфонамид (99 %), т. пл. 142 °C. ИК-спектр (v, см-1): 3335 (N-H); 1590 (C=О); 1360,1170 (SO2). Отделенный фильтрованием оксид алюминия обработан аммиачной водой, после упаривания воды получен трихлорацетат аммония (99 %). Использование Al2O3 как носителя воды и катализатора гидролиза позволило провести реакцию гидролитического расщепления сульфонамида (III а) в мягких условиях с количественными выходами указанных продуктов.
Для дальнейшего выявления синтетического потенциала 2,6-дизамещенных 1,4,3,5-оксатиадиазин-4,4-диоксидов (I) в настоящей работе было исследовано их взаимодействие с непредельными соединениями. В реакциях эквимолярных количеств диоксидов (I а-г) с алкенами и алкином (циклогексен, стирол, 3,3-диметилбутен-1, фенилацетилен) при нагревании (60–65 °C, 7часов) в бензоле получены насыщенные (IV а-в) и ненасыщенный (V) 1,4,5-оксатиазин-4,4-диоксиды. Для выделения соединений (IV а-в, V) из реакционной массы растворитель отгонялся в токе воздуха, остаток обрабатывался смесью гексан : диэтиловый эфир (2:1).
Выходы, температуры плавления, ИК-спектры и спектры ЯМР 1Н полученных соединений (IV а-в) и (V) приведены в табл. 3.
Таблица 3
Выходы, температуры плавления, ИК и ЯМР 1Н спектры 1,4,5-оксатиазин-4,4-диоксидов (IVа-в) и (V)
|
№ соединения |
Выход, % |
Т. пл., °C |
ИК-спектр, v, см-1 |
Спектр ЯМР 1Н, δ, м. д. |
||
|
SO2 |
С=С |
C=N |
||||
|
IVа |
94 |
129–130 |
1170, 1370 |
– |
1710 |
1,00–1,85 м (8H, CH2); 2,62 д (1H, OCH); 4,65 с (1Н, SCH) |
|
IVб |
88 |
142 |
1175, 1375 |
– |
1730 |
3,03–3,76 м (2Н, CН2); 5,64–6,26 м (1Н, СН); 6,50–8,00 м (5Н, С6Н5) |
|
IVв |
92 |
183 |
1175, 1350 |
– |
1695 |
1,09 с (9Н, (СН3)3С); 3,27 т (1Н, СН); 3,59 кв (1Н, СН); 4,74 кв (1Н, СН) |
|
V |
95 |
148 |
1175, 1370 |
1530, 1620 |
1740 |
6,69 с (1Н, С); 7,37–7,81 м (5Н, С6Н5) |
Ранее была показана возможность замещения иминного фрагмента в диоксидах (I), имеющих сильноакцепторные группы R1 и слабоакцепторные или донорные группы R2, при их взаимодействии с цианосодержащими соединениями, имеющими более нуклеофильно активные цианогруппы [2].
В настоящей работе при взаимодействии диоксидов (I) с ароматическими реагентами иминный фрагмент замещался на соответствующий ароматический с раскрытием исходного гетероцикла, а в реакциях с ненасыщенными реагентами – на этиленовый, с сохранением в продукте гетероциклической структуры. Следует отметить, что во всех сериях опытов с ароматическими и непредельными соединениями уходящей из гетероцикла (I) является нитрильная компонента с заместителем R2, более электронодонорным, чем заместитель R1.
Превращения диоксидов (I), сопровождающиеся уходом из гетероцикла нитрильной компоненты, можно представить как ступенчатый процесс, включающий образование промежуточных реакционных частиц-сульфониламидатов (II) на первой стадии и взаимодействие их с реагентом – на последующей, как показано выше на схеме. Подтверждением такого механизма реакции явилось образование N-ациларенсульфонамидов (III), насыщенных и ненасыщенных 1,4,5-оксатиазин-4,4-диоксидов (IV, V) в изученных в настоящей работе реакциях диазинов (I) c аренами, алкенами и фенилацетиленом соответственно. Образование соединений указанного класса (IV, V) было показано во взаимодействии алкенов и алкинов с системой 6-трихлорметил-1,3,2,4,5-диоксадитиазин-2,2,4,4-тетраоксид – пиридин, в которой действительно генерируются сульфониламидаты (II), строение которых доказано рентгеноструктурным анализом [10].
Однако в указанной авторами системе 1,4,5-оксатиазин-4,4-диоксиды (IV) всегда образовывались в смеси с 1,2-тиазетидин-1,1-диоксидами (в различных соотношениях и с невысокими выходами), что снижает синтетическую значимость описанных реакций. Использование же в реакциях с непредельными соединениями 2,6-дизамещенных 1,4,3,5-оксатиадиазин-4,4-диоксидов (I) позволяет осуществить целенаправленный синтез соединений (IV, V) с высокими выходами, как показано в настоящей работе.
Выводы
Таким образом установлено, что 2,6-дизамещенные 1,4,3,5-оксатиадиазин-4,4-диоксиды (I), имеющие несколько реакционных центров (чем определяется многовариантность возможных путей их реагирования и синтетическая значимость), перспективны и как структурные блоки в синтезах, включающих замещение нитрильной компоненты в гетероцикле на этиленовый, иминный, ароматический фрагменты с широким выбором заместителей.
Также показано, что эти реакции протекают через промежуточное образование реакционных частиц-сульфониламидатов.
Поскольку исследованные превращения реализуются узкоселективно, в большинстве случаев с практически количественными выходами продукта, они представляет интерес для целенаправленного получения циклических и ациклических сульфонилазотсодержащих соединений заданного строения.
Библиографическая ссылка
Сажина Е.Н. ВЗАИМОДЕЙСТВИЕ 2,6-ДИЗАМЕЩЕННЫХ 1,4,3,5-ОКСАТИАДИАЗИН-4,4-ДИОКСИДОВ С АРОМАТИЧЕСКИМИ И НЕПРЕДЕЛЬНЫМИ РЕАГЕНТАМИ // Международный журнал прикладных и фундаментальных исследований. 2018. № 11-2. С. 253-257;URL: https://applied-research.ru/en/article/view?id=12485 (дата обращения: 13.07.2026).