
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
MECHANISMS AND FACTORS OF ANGIOGENESIS

Ангиогенез (а) – сложный процесс формирования новых кровеносных сосудов в органах либо тканях, предопределенный строго поочередной работой множества факторов в пространственно-временной позиции. Новые капилляры образуются из мелких кровеносных сосудов, путем активизирования эндотелиальной клетки, преобразования в них протеиназ, деградации внеклеточной структуры ткани, разрастания и передвижения клеточных структур. Впоследствии благодаря формированию клетками первичных высокопроницаемых сосудов, начинается стабилизация и «взросление» структур клетки с помощью притягивания перикапиллярных клеток и клеток гладких мышц. В результате чего строится многогранная сеть сосудов. В норме ангиогенез происходит в спокойном темпе, активируется он при условии тканевых ранений, наличия тромбов и других патологических процессах [1]. Физиологический ангиогенез – это реакция ткани на гормональную стимуляцию (ангиогенез в репродуктивной системе) или изменения в окружающей среде (в ответ на ишемию ткань может расширять сосудистую сеть). Исследования последних научных работ приходят к выводу, что главным стимулом ангиогенеза является дефицит кислорода, который вызывает гипоксию или ишемию, при этом HIF-1 содействует выразительности васкулярных факторов, а именно фактору роста внешнего сосудистого слоя VEGF и его нервных окончаний, который является основой регулирования роста сосудов в различных периодах развития организма [2]. Физиологический ангиогенез представлен реакцией адаптации к дефициту кислорода, поскольку VGEF считается стресс-индуцированным белком, регулируемый глюкозой и кислородом. Фактор роста индивидуально отбирает эндотелиальные клетки (ЭК) для активации их преобразования и передвижения. Увеличивает пропускаемость сосуда для прохода белков в периваскулярное пространство, необходимое для контролируемой миграции ЭК и развитию вазодилатации [3]. В этапе развития новой сети сосудов принимает участие проангиогенный фактор, который сдерживает эндотелиальную пролиферацию, снижает проницаемость сосуда и содействует притягиванию перикапиллярных клеток. Tie2 (тирозинкиназные рецепторы) играют ведущую роль в процессах роста, развития и дифференцировки клеток. Они вместе с ангиопоэтинами играют роль в корректировке сопряжения эндотелия с рядом лежащими клетками [4]. Для роста системы сосудов в эмбриональном периоде необходима система сигналов Tie/Ang, соединяемая с VEGF и его рецепторами, равно как и каскад сигнализации. Tie2/Ang1 является несамостоятельным, промотирующим ассоциацию перицитов и эндотелия, снижающим сосудистую проницаемость и обладающим противовоспалительной активностью каскадом сигнализации [5]. Ang1 помогает образовывать связь между перицитами и эндотелиальными клетками при связывании с экспрессируемым на поверхности клеток эндотелия рецептором Tie2, помогая стабилизации, находящегося в стадии развития сосудистой системы [6, 7]. 2. Тромбоцитарный ФР (PDGF), который привлекает перициты и ГМК. Это белок, синтезируемый в мегакариоцитах и находящийся в гранулах тромбоцита. Все элементы – это результат роста фактора около тысячи молекул тромбоцитов. Фактор – мощный стимул восстановления тканей. Рецепторы для этого находятся в стенке сосудов на поверхности фибробласта и клетках гладкой мускулатуры. PDGF активизирует пролиферацию таких клеток. Более того, PDGF усиливает выработку компонентов соединительной ткани (коллагена, гистамина и др.) [8]. 3. Трансформирующий ФР-β1 (TGF-β1) стимулирует синтез белков внеклеточной матрицы. Контролирующий пролиферацию полипептид (представитель цитокинов) в большинстве клеток также регулирует дифференциацию и другие функциональные особенности. Члены семейства TGF-β1 проявляют множественное воздействие на огромное количество видов клеток и способствуют контролю роста клеток, дифференциации и апоптоза, а также в модуляции иммунной системы [9]. Артериогенез способствует формированию коллатеральных сосудов из неактивных артериальных сетей, по которым кровь проходит в места замыкания. Главным катализатором такого процесса является увеличение напряжения сдвига выше места окклюзии, способствующего преобразованию молекул адгезии клетками эндотелия с последующей аккумуляцией моноцитов в стенке сосуда. Они секретируют функционирующие ФР, основными регуляторами артериогенеза являются фактор роста фибробластов (FGF), и PDGF, VEGF и CXC-хемокины (подсемейства, характеризующиеся наличием одной аминокислоты, которая разделяет N-концевые цистеины) [10]. Действия ангиогенеза контролируются ФР во временном пространстве, этот факт следует учесть во время терапевтического ангиогенеза. Стабильное состояние сосудистой сети в организме постнатального периода обеспечивается равноценным соотношением между активаторами ангиогенеза (в основном ФР и цитокинами) и его ингибиторами (тромбоспондином, ангиостатин, тумастин, эндостатином и др.), движение такого баланса в сторону активаторов, в большинстве случаев, непродолжительный, ведет за собой активацию ангиогенеза [11]. Примерами являются воспаление, заживление ран, ишемия.
Этапы ангиогенеза.
Данный процесс и образование отростчатых сосудов проходит в несколько последовательных шагов. В первой фазе начинается активация перицитов, которые находятся в тесном контакте с эндотелием, они увеличиваются в объеме, укорачивая свои отростки. Таким образом, происходит ослабление межклеточных контактных соединений. Перициты проецируются в периваскулярное пространство, происходит деградация базальной мембраны и диссоциация перицитов и эндотелия [12]. Хотя процесс на начальных стадиях роста эндотелиоцитов в новообразованную васкуляризованную ткань может протекать без помощи перицитов, в последующей работе именно они локализуются по ходу прорастания эндотелия и формируют процессы, которыми направляются новообразованные сосуды [13]. Клетки эндотелиоциты берут начало своего роста в тканях по направлению Ang-1 продуцирующей ткани и, производя ферменты, катепсины и активаторы плазминогена, которые ведут к ухудшению базальной мембраны, так же матриксные металлопротеиназы (MMPs), являющиеся основными протеолитическими энзимами, принимающими участие в этом процессе. Внеклеточный матрикс, расщепляясь, формирует полипептиды, фрагменты этих белков имеют как про- так и антиангиогенные эффекты. Растворение белков внеклеточного матрикса проходит под контролем ингибиторов протеаз (UAP, PAI) [14]. При участии молекул клеточной адгезии и 17 интегринов, лигандами для которых служат белки внеклеточного матрикса (фибронектин, ламинин, витронектин), ослабление межклеточных контактных соединений эндотелиальных клеток и разрушение базальной мембраны дает начало далее следующему перемещению эндотелиальных клеток в околососудистый участок [15]. Клетки эндотелия начинают активно пролиферировать, сформировывая структуры в виде канала, далее преобразовываясь в зрелую сосудистую сеть. Некоторые сосуды микроциркуляторного русла объединяются в целостную сеть, посредством которой происходит перфузия тканей (рис. 1). До этого момента VEGF влияет на сохранение клеток эндотелия и их целостность [16].
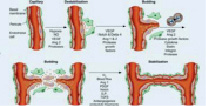
Рис. 1. Этапы ангиогенеза
Момент подъема эндотелия сосудов. В процессе проницаемости сосудов лежит база регулировки А. VEGF – мощнейший индуктор ангиогенеза в целом ряде опытных моделей in vivo [17]. Он представляет собой гомодимерный, высокогликолизированный, митогенный белок, предназначенный для эндотелиальных клеток. Большинство ученых считают, что VEGF взаимодействует с цитокинами, которые имеют сенсоры с протеолитическими ферментами и растворимые антагонисты. Они, взаимодействуя, регулируют высвобождение цитокинов из внеклеточного матрикса [18]. Группа VEGF включает в себя ряд образцов: EGF-A способствует наращиванию проницаемости сосуда; VEGF-B регулирует спад внеклеточного матрикса, адгезии и клеточного передвижения; VEGF-C и VEGF-D играют главную роль в регуляции лимфатических кровеносных сосудов; так же VEGF-E является вирусным гомологом и способствует плацентарному подъему (PIGF). Он отвечает за построение сосудистой сети в плаценте (рис. 2).
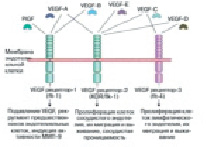
Рис. 2. Семейство VEGF
Совместное взаимодействие VEGF с рецепторами VEGFR-2 подключает активацию всех имеющихся сигнальных путей каскада. Два таких примера показаны на рисунке. Такое взаимодействие приводит к выживанию клетки и проницаемости сосудов, путем поглощения генов, которые способствуют пролиферации и передвижению клеток эндотелия. К примеру, связывание VEGF с сенсором VEGFR-2 приводит к димеризации сенсора с дальнейшей активацией пути синтеза ДНК и подъема клеток PLC-PKC-Raf-MEK-митогенактивированной белковой киназы (MAPK), а еще дальнейшей инициацией синтеза ДНК и подъема клеток, за это время как активация фосфатидилинозита 3’-киназы (PI3K)-Akt-way обязана прирастить подъем эндотелиальных клеток. Передвижение клеток и изменения в цитоскелете актина вызывает ген Src. Рецепторы VEGF располагаются на поверхности эндотелиальной клетки, но не теряют способности стать внутриклеточными [19]. Они являются участниками процесса приспособления клетки для выживания. VEGFR-2 представлен полноразмерным сенсором, прикрепляющимся к плоскости клеточки. VEGF-CcVEGFR-3 связывание опосредует лимфангиогенез. VEGF имеет возможность вязать рецепторный нейропилин (NRP), который имеет возможность работать как coreceptor с VEGFR-2 (горизонтальная стрелка 1) и имеет возможность регулировать A [20]. Комбинированное внедрение VEGF в экспериментальных исследовательских работах, а еще фактора стабилизации сосудов ангиопоэтин-1, и тромбоцитов FR (PDGF-BB) в композиции с FGF-2 вызывает возникновение сосудистой сети, которая продолжает оставаться размеренной сквозь 1 год впоследствии остановки данных моментов [21, 22]. Иным раскладом к более равновесной стимуляции ангиогенеза, вполне вероятно, может быть создание генетических структур, основанных на консистенции геномной ДНК, а еще cDNA-форм гена VEGF, имеющие в для себя экзоны и интроны в иной области слияния. Другая стратегия имеет возможность основываться на применении генов, которые кодируют моменты, которые активируют поглощение множества ангиогенных молекул [23]. Фактором, обеспечивающим больше обобщенные сигналы ангиогенеза, имеет возможность быть активатор плазминогена наподобие урокиназы (urokinase) – протеаза серина, ведущей регулятор внеклеточного протеолиза, а еще моделирование тканей. Урокиназа инициирует составление капилляров и артериол и наращивает скопление макрофагов в зоне периинфаркции, сокращаяет величину возникших, увеличивает васкуляризацию, готовит более скорым восстановление перфузии и не позволяет развиваться некрозу в ишемической конечности [24].
Заключение
Дефицитность кровоснабжения приводит к гипоксии по причине понижения диффузии воздуха. Гипоксия считается более необходимым катализатором A, есть активация метаболических стезей, которые индуцируются белками, этими как момент гипоксии 1, собственно, что приводит к наращиванию экспрессии проангиогенных моментов, этих как моменты подъема VEGF и фибробластов [25]. Впоследствии подключения А случается перелом соединительнотканной пластинки и внеклеточного матрикса (ВКМ), вследствие увеличенной энергичности матрикса. Далее клетки организуются в канальцы, с просветами образуя свежую капиллярную металлопротеиназу (ММП). Во время сего процесса притягиваются перициты, которые прикрепляются к свежим кровеносным сосудам и стабилизируются. До сего этапа созревания единство и выживание эндотелиальных клеток находятся в зависимости от VEGF1.13. Другой методикой экспрессии считается втягивание воспалительных клеток, цитокинов ФНО (фактор некроза α-опухолей) и ИЛ-1, которые в собственную очередь индуцируют продукцию обычных клеток. Подъем микрососудов продолжается до тех пор, пока же не достигнется очень максимально вероятная близость к клеточке. Впоследствии А. перебегает в стадию спокойствия (в дамской репродуктивной системе ангиогенный цикл считается исключением). Каждое наращивание массы ткани сопрягается с уноваскуляризацией, которая поддерживает необходимую плотность сосудов. Например, А. индуцируется, когда метаболическая надобность выше перфузионную дееспособность имеющих место быть сосудов. По-видимому, устройство данной адаптивной реакции заключается в том, собственно, что условный недостаток воздуха приводит к ужесточению ангиогенных стимулов.
Библиографическая ссылка
Шамитова Е.Н., Сымулова И.С., Леванова М.М., Кашеварова Э.А. МЕХАНИЗМЫ И ФАКТОРЫ АНГИОГЕНЕЗА // Международный журнал прикладных и фундаментальных исследований. 2019. № 9. С. 30-34;URL: https://applied-research.ru/en/article/view?id=12846 (дата обращения: 01.07.2026).