
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
SEARCH FOR COMPOUNDS WITH ANALGESIC ACTIVITY USING MOLECULAR DOCKING IN STRUCTURE – ACTIVITY STUDIES ON CYCLOOXYGENASE 1 AND 2 ENZYMES IN A SERIES OF N-SUBSTITUTED ANTHRANILIC ACIDS

Моделирование взаимодействия с организмом с целью проведения скрининга с использованием молекулярного докинга, и полученных на его основе соотношений «структура – активность» позволяет проводить целенаправленный поиск соединений с выбранным видом фармакологической активности. Использование молекулярного докинга возможно в том случае если известна мишень, с которой взаимодействует исследуемый лиганд. Перед проведением скрининга методом молекулярного докинга важно узнать фермент, воздействие на который приводит к появлению у исследуемого лиганда анализируемой биологической активности.
Антраниловая кислота принимает участие в широком спектре биохимических реакций, используется в качестве одного из ключевых реагентов в полусинтетическом методе получения ряда антибиотиков, как, например, кальцимицина, проявляющего противомикробную активность [1]. Производные антраниловой кислоты относятся к классу биологически активных соединений, обладающих широким спектром фармакологической активности: анальгетической [2], противовоспалительной и противомикробной [3–6].
Проведение моделирования по анальгетической активности является актуальным при проведении целенаправленного поиска веществ с выраженной анальгетической активностью. Сочетание молекулярного докинга с физико-химическими дескрипторами позволяет учитывать в проводимых исследованиях физико-химические свойства веществ, увеличивая качество модели, как, например, модель «структура – противовоспалительная активность» [7].
Цель исследования: исследование связи «структура – анальгетическая активность» с использованием молекулярного докинга по ферментам циклооксигеназа (ЦОГ) 1 и 2 в ряду N-замещенных антраниловых кислот (10 соединений)
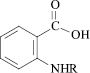
R = CH2CH=C(Cl)CH3 (1);
R = CH2CH=CH2 (2);
R = COC6H3(2,4-Cl2) (3);
R = COCONHCH2CH=CH2 (4);
R = COCOOC2H5 (5);
R = CO(2-фурил) (6);
R = CO Ad (7);
R = CH2C6H5 (8);
R = COC6H4(2-OCH3) (9);
R = COCH2C6H5 (10)
Материалы и методы исследования
Структуры всех лигандов были построены и оптимизированы с использованием программы Gaussian 03 полуэмпирическим методом РМ3 с полной оптимизацией геометрии молекул, и затем были конвертированы в 3D-формат (.pdb) с помощью программы ChemBio3D Ultra 12,0.
Определение анальгетической активности (АА) проводили на беспородных белых мышах массой 18–23 г на модели «горячей пластинки». Исследуемые соединения вводили внутрь в дозе 50 мг/кг за 0,5 ч до помещения животных на металлическую пластинку, нагретую до 53,5 °С. Каждое соединение испытывали на пяти животных. Показателем изменения болевой чувствительности служила измеряемая в секундах длительность пребывания животных на горячей пластине до момента облизывания ими задних лапок [8].
Моделирование лиганд – рецепторных взаимодействий осуществляли программой AutoDock 4.0 в составе программного комплекса MGL Tools 1.5.6, с использованием Ламарковского генетического алгоритма, который позволяет воспроизводить строение комплексов более точно. При проведении молекулярного докинга мы использовали трёхмерные модели молекул фермента, информация о которых получена из базы данных RCSB Protein Data Bank ЦОГ 1 (PDB ID code: 3N8X [9]) и 2 (PDB ID code: 1PXX [10]). Изначально, все молекулы воды были удалены из структуры белка. Файлы рецептора и лигандов были конвертированы в формат PDBQT–файла, с добавлением недостающих атомов водорода и частичных атомных зарядов по методу Гастейгера.
В процедуре докинга, с координатами точек (60×60×60) вокруг каждого моделируемого участка, по ферменту ЦОГ 1 при построении Grid–карт за центр были взяты координаты лиганда (x = –19,44, y = –54,38, z = 6,07), а для фермента ЦОГ 2 координаты лиганда (x = 26,25, y = 25,04, z = 14,66). В результате получено 10 Grid–карт по параметрам для исследуемых соединений по ЦОГ 1 и 2. Оценку качества позиционирования характеризовали величиной RMSD, представляющей собой среднеквадратичное отклонение положения лиганда после докинга от его начального положения в белке.
Результаты исследования и их обсуждение
Структуры всех лигандов были построены и оптимизированы с использованием программы Gaussian 03 полуэмпирическим методом РМ3 с полной оптимизацией геометрии молекул. В результате получены квантово-химические дескрипторы: суммарные значения напряженности электрического поля Σ(Е), потенциала Σ(φ) и заряда Σ (|q|) на атомах углерода, кислорода, азота и водорода (табл. 1). Рассчитанные квантово-химические дескрипторы использовали для теоретического расчёта физико-химических дескрипторов исследуемых соединений с помощью зависимостей «структура – свойство»: констант липофильности (log Pрассч) [11], констант кислотности (pKaрассч) и основности (pKврассч) [12] (табл. 1).
Значения констант липофильности лежат в интервале от 0,44 (у соединения 5) до 2,92 (соединение 6). Константы кислотности, характеризующие ионизацию, рассчитанные значения которых находятся в пределах от 5,10 (соединение 3) до 7,89 (для соединения 4). Значения констант основности, в интервале от 11,91 у соединения 5 до 13,42, для соединения 3.
Для 10 соединений из ряда N-замещенных антраниловых кислот была экспериментально определена АА. Установлено, что большинство изучаемых амидов обладает различным уровнем АА от 13,5 до 27 с (табл. 2).
Моделирование лиганд-рецепторных взаимодействий осуществляли программой AutoDock 4.0 в составе программного комплекса MGL Tools 1.5.6, с использованием Ламарковского генетического алгоритма, который позволяет воспроизводить строение комплексов более точно.
В качестве активного сайта фермента ЦОГ 1 был принят участок макромолекулы, содержащий аминокислоты (SER530, TYR385, ARG120, TYR355, ALA527, MET113, LEU531, VAL116, VAL349, ILE523, PHE518, LEU352, PHE381, TRP387, LEU359, ALA527, ILE517, LEU384) [9] и ЦОГ 2 (VAL349, ALA527, TYR355, VAL523, LEU352, TRP387, PHE518, LEU384, TYR385, ARG120, SER530) [10]. Значимыми считали гидрофобные взаимодействия и образование межмолекулярных водородных связей с аминокислотами активного участка ферментов ЦОГ 1 и 2.
Результат проведенного молекулярного докинга по ферментам ЦОГ 1 и 2 представлен в табл. 2 виде скоринговых функций: энергия связывания (Binding energy (BeЦОГ1 и BeЦОГ2)) и межмолекулярная энергия (Intermolecular energy (ImeЦОГ1 и ImeЦОГ2)), характеризующие взаимодействие лиганда с рецептором.
Таблица 1
Квантово-химические дескрипторы N-замещенных антраниловых кислот
|
№ |
C(E) |
N(E) |
C(φ) |
O(φ) |
N(φ) |
O(|q|) |
logPрассч |
pKaрассч |
pKврассч |
|
1 |
5,43 |
0,40 |
125,71 |
27,07 |
13,18 |
0,70 |
1,74 |
6,74 |
12,40 |
|
2 |
4,97 |
0,40 |
110,98 |
25,96 |
12,79 |
0,69 |
1,66 |
6,43 |
12,60 |
|
3 |
8,47 |
0,52 |
171,22 |
41,01 |
13,83 |
1,02 |
2,48 |
5,10 |
13,42 |
|
4 |
6,18 |
0,95 |
143,04 |
54,51 |
26,62 |
1,32 |
1,06 |
7,89 |
12,03 |
|
5 |
5,32 |
0,49 |
126,17 |
67,55 |
13,07 |
1,61 |
0,44 |
7,17 |
11,91 |
|
6 |
5,87 |
0,22 |
135,61 |
54,03 |
13,27 |
1,11 |
2,92 |
6,34 |
13,03 |
|
7 |
13,31 |
0,67 |
264,12 |
46,25 |
15,79 |
1,03 |
2,04 |
3,93 |
14,14 |
|
8 |
7,51 |
0,24 |
169,77 |
27,40 |
14,15 |
0,70 |
1,95 |
6,11 |
13,22 |
|
9 |
7,76 |
0,23 |
185,36 |
58,14 |
14,42 |
1,23 |
2,89 |
6,82 |
12,74 |
|
10 |
8,41 |
0,10 |
186,32 |
42,85 |
14,53 |
0,97 |
1,33 |
5,47 |
13,99 |
Таблица 2
ААэксп. и скоринговые функции по ЦОГ 1 и 2 N-замещенных антраниловых кислот
|
№ |
ААэксп., сек |
Beцог1 |
Imeцог1 |
Beцог2 |
Imeцог2 |
|
1 |
27,0 |
–6,12 |
–7,61 |
–4,87 |
–6,36 |
|
2 |
23,0 |
–4,15 |
–6,44 |
–4,62 |
–6,11 |
|
3 |
17,3 |
–6,91 |
–8,10 |
–6,24 |
–7,43 |
|
4 |
16,2 |
–5,99 |
–7,78 |
–5,06 |
–6,85 |
|
5 |
13,5 |
–5,42 |
–7,21 |
–4,69 |
–6,48 |
|
6 |
17,0 |
–6,18 |
–7,38 |
–5,27 |
–6,46 |
|
7 |
17,1 |
–6,71 |
–7,91 |
–7,14 |
–8,33 |
|
8 |
14,6 |
–7,39 |
–8,89 |
–6,20 |
–7,69 |
|
9 |
17,6 |
–6,35 |
–7,85 |
–6,42 |
–7,74 |
|
10 |
22,8 |
–6,35 |
–7,84 |
–6,68 |
–8,17 |
С целью составления модели «структура – анальгетическая активность» проведен множественный линейный регрессионный анализ с использованием программы Statistica 6, зависимости ААэксп. от констант липофильности (logPрассч), ионизации (pKaрассч и pKврассч) и скоринговых функций по ферментам ЦОГ 1 и 2 (BeЦОГ1, BeЦОГ2, ImeЦОГ1, ImeЦОГ2).
Всего было сгенерировано свыше 23 уравнений регрессии методом пошагового включения параметров с использованием программы Statistica 6, из которых были отобраны четыре уравнения 1–4 (табл. 3).
Таблица 3
Уравнения множественной регрессии зависимости ААэксп. от logPрассч, pKврассч, BeЦОГ1, ImeЦОГ1, BeЦОГ2, ImeЦОГ2.
|
№ |
Уравнение регрессии |
R |
F |
S |
Q2LOO |
|
1 |
ААрассч.1 = 43,689 – 3,519×BeЦОГ1 + 6,107×ImeЦОГ1 |
0,398 |
0,66 |
4,42 |
0 |
|
2 |
ААрассч.2 = –5,451 + 4,089×pKврассч + 4,784×BeЦОГ1 |
0,880 |
7,09 |
2,69 |
0,40 |
|
3 |
ААрассч.3 = 21,906 +2,649×pKврассч + 4,946×ImeCOX1 |
0,745 |
4,36 |
3,21 |
0,04 |
|
4 |
ААрассч.4 = –20,489 + 4,501×pKврассч + 7,190×BeЦОГ1 – 3,187×ImeЦОГ1 |
0,899 |
8,46 |
2,27 |
0,51 |
Таблица 4
log Pрассч, pKaрассч, pKврассч, скоринговые функции по ЦОГ 1 и 2, ААрассч.4, ААэксп. N-замещенных антраниловых кислот (10 соединений)
|
№ |
log Pрассч |
pKaрассч |
pKврассч |
Beцог1 |
Imeцог1 |
Beцог2 |
Imeцог2 |
ААрассч.4 |
ААэксп.,сек |
|
1 |
2,50 |
6,00 |
12,72 |
–6,95 |
–8,12 |
–6,54 |
–7,74 |
13,6 |
11,7 |
|
2 |
1,57 |
7,11 |
11,86 |
–5,10 |
–8,12 |
–6,15 |
–7,39 |
11,1 |
13,8 |
|
3 |
1,02 |
6,23 |
12,86 |
–5,63 |
–6,82 |
–4,99 |
–6,18 |
18,7 |
21,3 |
Установлено, что полученное уравнение 4 имеет наибольшие значения коэффициента корреляции (R), критерия Фишера (F) и минимальное значение среднеквадратичной ошибки (S). Проведена оценка составленных уравнений методом перекрестного контроля с выбором по одному (Leave-one-out Cross-validation, LOO) (Q2LOO), с использованием программы Statographics. Определен коэффициент детерминации предсказаний Q2LOO, показывающий значимость составленных уравнений. К наиболее значимым по рассчитанным критериям R, F, Q2LOO относится модель в виде уравнений 4.
Уравнение 4, апробировано для теоретического расчёта АА на трех примерах соединений 11 – 13 из ряда N-замещенных антраниловых кислот (табл. 4).
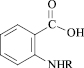
R = COC6H3(3-CH3) (11);
R = CO C6H4 (4-NO2) (12);
R = COCH2Cl (13)
Для проверки проведенного прогноза по соединениям 11–13, была экспериментально определена АА (ААэксп.) (табл. 4).
Оценку точности прогнозирования АА по полученному уравнению 2 проводили с использованием среднеквадратичной ошибки прогноза (Sпрогн.).
Заключение
Проведен теоретический расчёт физико-химических дескрипторов исследуемых соединений с использованием зависимостей «структура – свойство». Для 13 соединений из ряда N-замещенных антраниловых кислот была экспериментально определена АА.
Выполнены исследования взаимодействия анализируемых производных с ЦОГ 1 и 2 методом молекулярного докинга, в результате получены скоринговые функции BeЦОГ1, BeЦОГ2, ImeЦОГ1, ImeЦОГ2. Составлена модель «структура – анальгетическая активность». Проведена проверка полученной модели на трех соединениях. Для оценки качества прогноза рассчитано значение Sпрогн., которое равно 2,43. Полученный результат сопоставим со значением S по уравнению 4.
Таким образом, можно сделать вывод, что полученную модель в виде уравнения 4 можно использовать для прогнозирования АА N-замещенных производных антраниловых кислот.
Библиографическая ссылка
Андрюков К.В., Коркодинова Л.М. ПОИСК СОЕДИНЕНИЙ С АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ С ИСПОЛЬЗОВАНИЕМ МОЛЕКУЛЯРНОГО ДОКИНГА В ИССЛЕДОВАНИЯХ «СТРУКТУРА – АКТИВНОСТЬ» ПО ФЕРМЕНТАМ ЦИКЛООКСИГЕНАЗА 1 И 2 В РЯДУ N-ЗАМЕЩЕННЫХ АНТРАНИЛОВЫХ КИСЛОТ // Международный журнал прикладных и фундаментальных исследований. 2019. № 12-1. С. 60-64;URL: https://applied-research.ru/en/article/view?id=12954 (дата обращения: 04.07.2026).