
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
THE PROMISING METHOD OF INTRODUCTION OF BIOLOGICALLY AKTIVE SUBSTITUENTS IN THE 1,4,3,5-OXATHIADIAZINE CYCLE

Памяти профессора Александра Андреевича Мичурина
Ранее [1–3] нами было показано, что гетероциклические сульфонилазотсодержащие соединения имеют большой препаративный интерес в связи с исключительной многовариантностью путей превращения одних и тех же исходных структур в разных условиях.
Также показано, что сульфонилазотсодержащие соединения, как ациклические, так и гетероциклические, обладают высокой биологической активностью и применяются в медикаментозной практике [4, 5], используются в качестве пестицидов и фунгицидов нового поколения [6, 7].
Однако применяемые в таких производствах способы получения высокозатратны и небезопасны в экологическом аспекте. Прямым взаимодействием триоксида серы и цианосодержащих соединений крайне трудно, а в ряде случаев невозможно осуществить направленные синтезы сульфонилазотсодержащих структур вследствие низкой селективности реакций, протекающих в таких системах. Поэтому важной проблемой современной органической химии является разработка новых, более простых и доступных путей синтеза сульфонилазотсодержащих циклических и ациклических соединений заданной структуры.
Метод предварительного перевода SO3 в связанные формы – аддукты и комплексы SO3 с нитрилами – значительно повышает избирательность их превращений и позволяет реализовать пути реагирования, абсолютно не характерные для непосредственного взаимодействия SO3 с цианосодержащими соединениями [1, 2].
В этом отношении перспективными субстратами показали себя 2,6-дизамещенные 1,4,3,5-оксатиадиазин-4,4-диоксиды (I). Наличие нескольких реакционных центров в молекуле определяет многообразные как уже подтвержденные, так и потенциально возможные пути их реагирования.
В продолжение проводимых ранее исследований интересным представляется изучение направленного введения в 1,4,3,5-оксатиадиазиновый цикл пиперидиновых и морфолиновых заместителей. Пиперидиновый цикл является одним из наиболее часто встречающихся фрагментов в природных и синтетических биологически активных веществах. Этим обусловлен интерес к химии самого пиперидина и его N-замещенных производных. Имеются сведения [8], что сульфонилазотсодержащие соединения именно с морфолиновым фрагментом успешно используются как лекарственные средства анальгетического действия.
Цель исследования – изучение возможности направленного введения в цикл 1,4,3,5-оксатиадиазин-4,4-диоксидов (I) заместителей, имеющих потенциальную биологическую активность.
Материалы и методы исследования
В исследовании рассмотрены 1,4,3,5-окса- тиадиазин-4,4-диоксиды с различными заместителями в гетероцикле, электронные свойства которых варьировались от сильно акцепторных до донорных (R1 = C6F5, CCl3, CBr3; R2 = C6H5, CH3, 4-ClC6H4, 2,4-Cl2C6H3). В роли цианосодержащих реагентов, включающихся в гетероциклы субстратов как новые иминные фрагменты с заданными заместителями, в работе рассмотрены N-цианопиперидин и N-цианоморфолин.
Индивидуальность полученных соединений и степень протекания реакции контролировалась методом тонкослойной хроматографии на пластинках Silufol UV-254, элюент – ацетон-гексан, 1:1 по объему, проявление осуществлялось парами йода.
Состав и строение синтезированных в работе соединений установлены с помощью элементного анализа, ИК- и ЯМР 1Н спектров. ИК-спектры соединений записаны на спектрофотометре Specord 80-M в метиленхлориде. Спектры ЯМР 1Н записаны на спектрометре Gemini 300 (рабочая частота 300 МГц) в (CD3)2CO, внутренний стандарт – ТМС.
Результаты исследования и их обсуждение
Нами было установлено, что процесс направленного замещения исходного иминного фрагмента цикла на иминный фрагмент с пиперидиновым или морфолиновым заместителем характерен только для асимметричных диоксидов (I), обязательно имеющих сильноакцепторные группы R1 (R1 = C6F5, CCl3, CBr3), в то время как второй заместитель R2 является электронодонором или слабым акцептором (R2 = C6H5, CH3, 4-ClC6H4, 2,4-Cl2C6H3).
Эта перециклизация реализуется при взаимодействии указанных диоксидов (I) с выбранными цианосодержащими реагентами, имеющими сильно нуклеофильные цианогруппы, и идет по схеме:
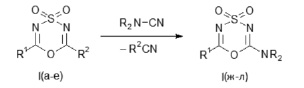
I, R1 = CCl3; R2 = CH3 (а); R1 = CBr3; R2 = CH3 (б); R1 = C6F5; R2 = CH3 (в);
R1 = CCl3; R2 = C6H5 (г); R1 = CCl3; R2 = 4-ClC6H4 (д); R1 = CCl3; R2 = 2,4-Cl2C6H3 (е);
I (R2 = NR2), R1 = CCl3; NR2 = N(CH2)5 (ж); R1 = CBr3; NR2 = N(CH2)5 (з); R1 = C6F5; NR2 = N(CH2)5 (и);
R1 = CCl3; NR2 = N(CH2)4О (к); R1 = CBr3; NR2 = N(CH2)4О (л).
Цианогруппы реагентов (N-цианопи- перидин, N-цианоморфолин) обладают очень высокой нуклеофильностью, поэтому можно предположить, что рассматриваемая реакция протекает по типу замещения, через координирование атома азота цианогруппы реагента и атома серы в субстрате (I).
Возможность образования такого типа соединений предполагалась ранее, но ни одно подобное соединение выделено не было. Таким образом, найденная реакция имеет высокую практическую значимость как удобный и единственный способ синтеза подобного типа 2,6-дизамещенных-1,4,3,5-оксатиадиазин-4,4-диоксидов с пиперидиновым или морфолиновым заместителями в гетероцикле (I, R2 = NR2).
Предложенный метод направленного введения в 1,4,3,5-оксатиадиазиновый цикл фрагмента с заданным заместителем возможно оптимизировать в плане снижения затрат и эффективности очистки получаемых продуктов. Поскольку обязательное условие протекания описываемого процесса – асимметричность гетероцикла в отношении электронных свойств двух его заместителей, и при этом реакция протекает однотипно в широком ряду исследованных субстратов (I, а-е), легко подобрать наиболее доступные диоксиды (I), способные к вовлечению в реакцию замещения иминного фрагмента.
В этом отношении видится целесообразным в качестве исходных структур использование диоксидов (I, а-в) с ацетонитрильным фрагментом в цикле. Кроме того, что ацетонитрил – это многотоннажный продукт и поэтому вполне доступен, он легко отделяется от синтезируемых диоксидов (I, R2 = NR2), что очень существенно для выделения и очистки последних.
В настоящей работе взаимодействием N-цианопиперидина и N-цианоморфолина с диоксидами (I, а-е) в соотношении, близком к эквимолярному, получена серия новых диоксидов (I, ж-л), в структуру которых направленно введены пиперидиновый или морфолиновый заместители.
Превращения проводились при 60 °С, в качестве растворителя использовался бензол. Оптимальная температура подбиралась опытным путем. Как показало наблюдение за ходом реакции методом тонкослойной хроматографии, понижение температуры кинетически тормозит реакцию и время превращения увеличивается, что экономически и технологически нецелеообразно. При повышении температуры на 5–7 °С не наблюдалось выраженного ускорения реакции, однако дальнейшее повышение температуры синтеза до 75–80 °С вызвало существенное нарастание цветности реакционной массы и образование неидентифицированных побочных продуктов, что регистрировалось хроматографически.
Продолжительность выдерживания реакционной массы при оптимальной температуре 60 °С варьировалась от 6 до 12 ч, при этом выходы продуктов приближались к количественным.
По истечении реакции, ход которой контролировался хроматографически, растворитель отгонялся вакуумированием реакционной массы, остаток после вакуумирования далее несколько раз обрабатывался промывкой гексаном и перекристаллизовывался из смеси растворителей метиленхлорид : гексан = 1 : 1. Из гексанового экстракта после отдувки растворителя количественно выделены соответствующие нитрилы R2CN, что также является одним из доказательств состава полученных диоксидов (I, ж-л).
Характеристики полученных диоксидов (I, ж-л) приведены в табл. 1.
Таблица 1
Продолжительность реакции, выходы, температуры плавления и ИК-спектры диоксидов (I, ж-л)
|
№ сое- динения |
Время, ч (60 °C, C6H6) |
Выход, % |
Т. пл. (разл.), °C |
ИК-спектр, v, см-1 |
|
|
SO2 |
C=N |
||||
|
Iж* |
12 |
99 |
144 |
1175, 1385 |
1685, 1745 |
|
Iз |
6 |
94 |
152 |
1165, 1375 |
1670, 1730 |
|
Iи |
8 |
93 |
138 |
1180, 1350 |
1665, 1730 |
|
Iк** |
7 |
82 |
173 |
1190, 1380 |
1665,1730 |
|
Iл |
7 |
93 |
178 |
1185,1360 |
1650, 1720 |
Примечания:
*Соединение (Iж) было получено по аналогичной методике взаимодействием N-цианопиперидина с диоксидом (Iг) с выходом 97 %, диоксидом (Iд) с выходом 98 % и диоксидом (Iе) с выходом 98 %.
**Соединение (Iк) получено по аналогичной методике взаимодействием N-цианоморфолина с диоксидом (Iе) с выходом 80 %.
Выходы в табл. 1 указаны для синтезов, в которых исходным субстратом являлся диоксид (Iа), преимущества использования которого были перечислены выше. Выходы диоксидов (I, R2 = NR2), полученных из других исходных субстратов исследуемой серии, даны в примечаниях к табл. 1.
Поскольку характеристические полосы поглощения в ИК-спектрах позволяют надежно судить только о присутствии в структуре соединений групп SO2 и C=N, в обязательном порядке проведен анализ спектров ЯМР 1Н, зафиксировавших резонансные сигналы входящих в состав заместителей протонов.
Данные ЯМР 1Н-спектроскопии и элементного анализа приведены в табл. 2.
Таблица 2
Данные элементного анализа диоксидов (I, ж-л)
|
№ сое- динения |
Спектр ЯМР 1Н, δ, м. д. (ДМСО-d6) |
Найдено/вычислено, % |
Формула |
||||
|
С |
Н |
Hlg |
N |
S |
|||
|
Iж |
1.48-1.83 м [6Н, СН2], 3.44-3.82 т [4Н, (СН2)2N] |
29.02 28.76 |
3.04 3.01 |
29.63 31.71 |
12.44 12.57 |
9.71 9.59 |
C8H10Cl3N3O3S |
|
Iз |
1.75-1.99м [6Н, СН2], 3.50-3.81 т [4Н, (СН2)2N] |
20.74 20.53 |
2.17 2.15 |
51.13 51.22 |
8.81 8.97 |
6.81 6.85 |
C8H10Br3N3O3S |
|
Iи |
1.56-1.70 м [6Н, СН2], 3.50-3.65 т [4Н, (СН2)2N] |
40.80 40.74 |
2.60 2.63 |
24.72 24.78 |
10.75 10.96 |
8.32 8.36 |
C13H10F5N3O3S |
|
Iк |
3.40-3.63 м [8H, CH2] |
25.01 24.98 |
2.47 2.39 |
31.72 31.60 |
12.53 12.49 |
9.58 9.53 |
C7H8Cl3N3O4S |
|
Iл |
3.52-3.64 м [8H, CH2] |
17.95 17.90 |
1.70 1.72 |
51.10 51.04 |
8.98 8.94 |
6.79 6.83 |
C7H8Br3N3O4S |
Выводы
Работа является продолжением исследований синтетических возможностей 2,6-дизамещенных 1,4,3,5-оксатиадиазин-4,4-диоксидов, имеющих несколько реакционных центров в молекуле. Установлена возможность направленного введения пиперидинового и морфолинового заместителей, обладающих потенциальной биологической активностью, в 1,4,3,5-оксатиадиазиновый гетероцикл с сохранением его структуры.
Показано, что предложенный в работе метод легко реализуется при наличии в исходном субстрате заместителей, существенно различающихся электронными свойствами, а именно – сильного акцептора R1 (который остается в составе исходного гетероцикла) и донорного или слабоакцепторного заместителя R2 (который в рассмотренном процессе в составе иминного фрагмента замещается).
Метод относительно гибок в варьировании уходящего нитрильного фрагмента, и это позволяет выбрать оптимальную исходную структуру. В работе исследованы превращения целого ряда исходных диоксидов (I) для выявления границ применимости предложенного метода. По доступности и простоте отделения от продукта оптимальным предложен диоксид с ацетонитрильным уходящим фрагментом.
Возможность существования синтезированных в работе соединений предполагалась ранее, но получены и выделены они не были. Поэтому найденная реакция имеет высокую практическую значимость не только как удобный, но на данный момент и единственный способ синтеза подобного типа 2,6-дизамещенных-1,4,3,5-оксатиадиазин-4,4-диоксидов с пиперидиновым или морфолиновым заместителями в гетероцикле.
Синтетическая значимость представленного в работе метода усиливается узкой селективностью описанных превращений и возможностью получать продукты с практически количественными выходами.
Библиографическая ссылка
Сажина Е.Н. ПЕРСПЕКТИВНЫЙ МЕТОД ВВЕДЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ЗАМЕСТИТЕЛЕЙ В 1,4,3,5-ОКСАТИАДИАЗИНОВЫЙ ЦИКЛ // Международный журнал прикладных и фундаментальных исследований. 2021. № 5. С. 67-70;URL: https://applied-research.ru/en/article/view?id=13221 (дата обращения: 01.07.2026).