
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
CLASSIFICATION QSAR MODELS OF SUBSTRATE ACTIVITY OF CHEMICAL RELATIVE TO P-GLYCOPROTEIN BASED ON THE SPECTRUM OF INTERATOMIC INTRAMOLECULAR INTERACRTION

Важной проблемой в поиске лекарств, действующих на мишени в пределах центральной нервной системы (ЦНС), является оценка возможности проникновения соединений через гематоэнцефалический барьер (ГЭБ). Этому процессу препятствует взаимодействие веществ с P-гликопротеином (P-gp), который является существенной частью ГЭБ. Этот трансмембранный белок выталкивает чужеродные вещества, токсины, а также лекарственные препараты за пределы ЦНС, обеспечивая тем самым стабильность среды. Однако некоторые молекулы могут модифицировать работу Р-gp, блокируя или существенно снижая его активность. Создание QSAR (Количественная Связь Структура Активность) моделей целенаправленного воздействия веществ на активность Р-гликопротеина является задачей многих исследований в области компьютерного поиска новых лекарственных препаратов [1]. При этом имеющиеся модели всё еще не обладают высокой точностью, трудны в интерпретации, не в полной мере учитывают физико-химические свойства атомов, особенно в части их способности к образованию водородных связей, а также особенности пространственного строения молекул.
В нашей лаборатории работы по компьютерному моделированию проникновения веществ из кровотока в мозг ведутся с 2011 г. В результате был создан ряд моделей проницаемости веществ через гематоэнцефалический барьер, базирующихся на различных QSAR методах.
Настоящее исследование посвящено созданию классификационных моделей субстратной специфичности веществ по отношению к Р-гликопротеину на базе новых пространственных (3D) дескрипторов и известных математических методов: линейного дискриминантного анализа (LDA), случайного леса (RF) и опорных векторов (SVM).
Материалы и методы исследования
Выборка соединений была сформирована на основе данных работы [2]. Для этого было отобрано 125 неионизированных (при pH = 7,0) молекул. В табл. 1 приведены вещества и их субстратная активность по отношению к Р-гликопротеину.
В качестве меры субстратной активности веществ были использованы величины ER (Efflux ratio), которые представляют собой отношение коэффициентов проницаемости соединений через клеточную мембрану в двух противоположных направлениях. Чем выше ER, тем выше концентрация вещества за пределами ЦНС. Все соединения были поделены на два класса. К первому классу (условно несубстраты) отнесли соединения с ER меньше 2. Ко второму классу (субстраты) были отнесены вещества с ER равным или превышающим 2. Таким образом, была сформирована выборка, состоящая из 69 несубстратов и 56 субстратов (табл. 1).
Для каждой молекулы был проведен полный конформационный анализ с использованием программы Cache Worksystem Pro 6.0 [3] (процедура Conflex). Использовали метод молекулярной механики в параметризации MM2 с порогом выхода из итерационного процесса минимизации энергии в 0,01 ккал/моль. Пространственные структуры с минимальной энергией напряжения использовали далее в качестве наиболее вероятных структур.
В качестве дескрипторов применяли интегралы интенсивностей спектров межатомных внутримолекулярных взаимодействий (СМВВ) в диапазоне от 0 до 20 ангстрем с шагом 0,2 ангстрема. Расчет СМВВ осуществляли с помощью программы MOLTRA [4]. Алгоритм программы разработан на основе модификации функции радиального распределения [5]. Пример расчета СМВВ представлен на рис. 1.
Каждый отдельный дескриптор представлял собой площадь прямоугольника с основанием 0,2 ангстрема (по оси абсцисс) и стороной (по оси ординат), равной величине интенсивности взаимодействия. Общее число дескрипторов на выбранном диапазоне составляло 100 для каждого типа взаимодействия. Учитывались следующие типы атом-атомных взаимодействий (ААВ): 1) взаимодействие атомов – доноров водородной связи (dd); 2) взаимодействие атомов – акцепторов водородной связи (aa); 3) взаимодействие атомов – доноров с атомами – акцепторами Н-связи (da); 4) взаимодействие положительно заряженных атомов (pp); 5) взаимодействие отрицательно заряженных атомов (nn); 6) взаимодействие положительно и отрицательно заряженных атомов (pn); 7) стерическое (ван-дер-ваальсовые) взаимодействие атомов (vdw). Таким образом, индивидуальный дескриптор представлял собой сумму ААВ определённого типа для всех атомных пар, находящихся на определённом расстоянии внутри молекулы. При отсутствии на данном расстоянии взаимодействующих пар атомов дескриптор был равен нулю.
Для бинарной классификации соединений (субстрат/несубстрат) применяли следующие методы машинного обучения: метод линейного дискриминантного анализа (LDA), метод случайного леса (RF) и метод опорных векторов (SVM). Линейный дискриминантный анализ проводили с использованием компьютерной программы SVD [6]. Для классификации на основе метода RF применяли программу rf5new [7] с исходными параметрами и числом деревьев, равным 100. Генерирование классификационных SVM моделей проводили с помощью компьютерной программы flssvm [8] на основе радиальной базисной функции.
Бинарные классификационные модели оценивали на основе следующих величин: TP – число правильно распознанных соединений, принадлежащих к первому классу, FN – число ошибочно распознанных соединений среди первого класса, TN – число правильно распознанных соединений, принадлежащих ко второму классу и FP – число ошибочно распознанных соединений среди второго класса.
Таблица 1
Соединения и их субстратная активность (ER)
|
№ |
Соединение |
ER |
№ |
Соединение |
ER |
№ |
Соединение |
ER |
|
1 |
Caffeine |
0.6 |
43 |
Bromazepam |
1.2 |
85 |
Lovastatinacid |
3.2 |
|
2 |
Testosterone |
0.7 |
44 |
Nitrazepam |
1.2 |
86 |
Mitoxantrone |
3.4 |
|
3 |
Diazepam |
0.7 |
45 |
Pyridostigmine |
1.2 |
87 |
Bromocriptine |
3.5 |
|
4 |
Methotrexate |
0.7 |
46 |
Nifedipine |
1.3 |
88 |
Cimetidine |
3.5 |
|
5 |
Indomethacin |
0.7 |
47 |
Amitriptyline |
1.3 |
89 |
Reserpine |
3.7 |
|
6 |
Antipyrine |
0.8 |
48 |
Clemastine |
1.3 |
90 |
Prednisone |
3.8 |
|
7 |
Haloperidol |
0.8 |
49 |
Solifenacin |
1.3 |
91 |
Prazosin |
3.8 |
|
8 |
Desipramine |
0.8 |
50 |
Clemastine |
1.3 |
92 |
Darifenacin |
4.0 |
|
9 |
Selegiline |
0.8 |
51 |
Clonazepam |
1.3 |
93 |
Lopinavir |
4.9 |
|
10 |
Lidocaine |
0.8 |
52 |
Ropinirole |
1.3 |
94 |
Aprepitant |
5.5 |
|
11 |
Amantadine |
0.9 |
53 |
Sumatriptan |
1.4 |
95 |
Prednisolone |
5.8 |
|
12 |
Carbamazepine |
0.9 |
54 |
Loratadine |
1.4 |
96 |
Vincristine |
6.3 |
|
13 |
Diphenhydramine |
0.9 |
55 |
Alprazolam |
1.4 |
97 |
Apomorphine |
6.9 |
|
14 |
Lamotrigine |
0.9 |
56 |
Zaleplon |
1.4 |
98 |
Sertraline |
7.4 |
|
15 |
Buspirone |
0.9 |
57 |
Oxcarbazepine |
1.4 |
99 |
Loperamide |
9.0 |
|
16 |
Nordazepam |
0.9 |
58 |
Reboxetine |
1.4 |
100 |
Desloratadine |
9.1 |
|
17 |
Mannitol |
0.9 |
59 |
Cyclobenzaprine |
1.4 |
101 |
Doxorubicin |
9.2 |
|
18 |
Enoxacin |
0.9 |
60 |
Tolterodine |
1.4 |
102 |
Digoxin |
10.6 |
|
19 |
Ciprofloxacin |
0.9 |
61 |
Tiagabine |
1.5 |
103 |
Clomipramine |
10.9 |
|
20 |
Zolpidem |
1.0 |
62 |
Riluzole |
1.5 |
104 |
Dexamethasone |
12.4 |
|
21 |
Zonisamide |
1.0 |
63 |
Diltiazem |
1.6 |
105 |
Daunorubicin |
14.2 |
|
22 |
Meprobamate |
1.0 |
64 |
Fluvoxamine |
1.7 |
106 |
Cabergoline |
14.3 |
|
23 |
Galantamine |
1.0 |
65 |
Nalbuphine |
1.7 |
107 |
Methylprednisolone |
14.7 |
|
24 |
Nortriptyline |
1.0 |
66 |
Risperidone |
1.8 |
108 |
Colchicine |
15.0 |
|
25 |
Phenytoin |
1.0 |
67 |
Duloxetine |
1.8 |
109 |
Nelfinavir |
15.6 |
|
26 |
Flumazenil |
1.0 |
68 |
Quetiapine |
1.8 |
110 |
Quinidine |
16.8 |
|
27 |
Naltrexone |
1.0 |
69 |
Pravastatinacid |
1.9 |
111 |
Etoposide |
18.8 |
|
28 |
Clonidine |
1.0 |
70 |
Simvastatinacid |
2.0 |
112 |
Indinavir |
22.5 |
|
29 |
Rivastigmine |
1.0 |
71 |
Olanzapine |
2.0 |
113 |
Dipyridamole |
22.7 |
|
30 |
Ropivacaine |
1.0 |
72 |
Neostigmine |
2.0 |
114 |
Erythromycin |
24.7 |
|
31 |
Ketoconazole |
1.0 |
73 |
Escitalopram |
2.0 |
115 |
Amprenavir |
27.4 |
|
32 |
Ramelteon |
1.0 |
74 |
Ranitidine |
2.1 |
116 |
Clarithromycin |
31.3 |
|
33 |
Imipramine |
1.1 |
75 |
Astemizole |
2.2 |
117 |
Eletriptan |
32.9 |
|
34 |
Maprotiline |
1.1 |
76 |
Ziprasidone |
2.3 |
118 |
Ximelagatran |
33.0 |
|
35 |
Topiramate |
1.1 |
77 |
Chlorpromazine |
2.4 |
119 |
Vinblastine |
37.3 |
|
36 |
Rosuvastatin |
1.1 |
78 |
Zolmitriptan |
2.5 |
120 |
Ritonavir |
38.8 |
|
37 |
Nalmefene |
1.1 |
79 |
Atomoxetine |
2.5 |
121 |
Domperidone |
47.0 |
|
38 |
Metoclopramide |
1.1 |
80 |
Clozapine |
2.5 |
122 |
Paclitaxel |
75.1 |
|
39 |
Sulfasalazine |
1.1 |
81 |
Trimethoprim |
2.8 |
123 |
Hoechst 33342 |
92.4 |
|
40 |
Tacrine |
1.1 |
82 |
Methysergide |
3.1 |
124 |
Rhodamine 123 |
115.0 |
|
41 |
Doxepin |
1.2 |
83 |
Paroxetine |
3.1 |
125 |
Saquinavir |
162.9 |
|
42 |
Promazine |
1.2 |
84 |
Famciclovir |
3.2 |
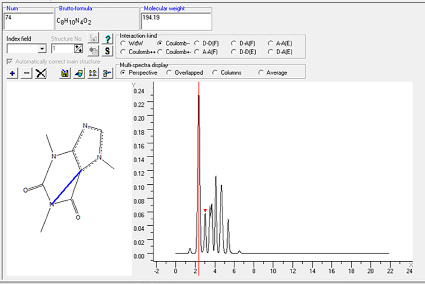
Рис. 1. Спектр межатомных внутримолекулярных взаимодействий отрицательно заряженных атомов кофеина
С использованием величин TP, FN, TN и FP рассчитывали следующие статистики:
чувствительность SN=TP/(TP+FN);
специфичность SP=TN/(TN+FP);
общую точность
AC=(TP+TN)/(TP+FN+TN+FP)
и коэффициент корреляции Мэтьюза: MCC=(TP*TN–FP*FN)/((TP+FP)*
*(TP+FN)*(TN+FP)*(TN+FN))0,5.
Тестирование полученных классификационных моделей проводили с помощью внутренней и внешней валидации. Для внутренней валидации использовали метод перекрестного контроля с выбором по пять. В методе RF аналогичную роль выполняла процедура OOB (out-of-bag). Для проведения внешнего тестирования общая выборка (125 соединений) была случайным образом разбита на обучающий ряд (100) соединений и тестовый ряд (25 соединений).
С целью уменьшения размерности пространства дескрипторы анализировали на основе алгоритма [9] с применением в качестве граничного значения величины коэффициента линейной парной корреляции, равной 0.8–0.9. В результате удалось сократить размер пространства со 100 до 12–61 дескриптора. Дальнейший отбор дескрипторов в классификационные модели осуществляли с использованием метода полного перебора комбинаций из 3–4-х дескрипторов. В качестве критерия отбора применяли модифицированную сбалансированную точность MBA= w1*(SN+SP)/2+w2*(1-abs(SN-SP)/2), где весовые коэффициенты w1 и w2 были равны 0,5.
Для оценки области применимости (AD) QSAR моделей использовали интервальный X-метод, суть которого состоит в определении попадания дескрипторов соединений тестовой выборки в соответствующие интервалы переменных для соединений обучающей выборки.
Результаты исследований и их обсуждение
В табл. 2 представлены характеристики моделей бинарной классификации, построенные на базе дескрипторов СМВВ.
Полученные данные свидетельствуют о том, что две модели (15, 21) имеют удовлетворительные (SN >= 0,75, SP >= 0,75 при внутренней и внешней валидации), статистические параметры, а три модели (3, 6, 12) – «почти» удовлетворительные. Обращает на себя внимание тот факт, что все вышеперечисленные модели созданы с использованием метода SVM. Очевидно, что по сравнению с другими методами (LDA, RF) он является наиболее адекватным.
Таблица 2
Классификационные модели субстратной активности молекул по отношению к Р-гликопротеину, рассчитанные на основе СМВВ дескрипторов
|
№ |
Тип |
Метод |
Дескрипторы |
Кросс-валидация (n = 100) |
Тестовая выборка (n = 25) |
||||||
|
SN |
SP |
AC |
MCC |
SN |
SP |
AC |
MCC |
||||
|
1 |
aa |
LDA |
17; 26; 28 |
0,836 |
0,578 |
0,720 |
0,432 |
0,929 |
0,455 |
0,720 |
0,445 |
|
2 |
aa |
RF |
7; 13; 44 |
0,782 |
0,711 |
0,750 |
0,494 |
0,714 |
0,727 |
0,720 |
0,439 |
|
3 |
aa |
SVM |
38; 51; 60 |
0,782 |
0,756 |
0,770 |
0,536 |
0,857 |
0,727 |
0,800 |
0,592 |
|
4 |
da |
LDA |
7; 25; 55 |
0,927 |
0,577 |
0,770 |
0,548 |
0,857 |
0,545 |
0,720 |
0,428 |
|
5 |
da |
RF |
27; 30; 70 |
0,727 |
0,733 |
0,730 |
0,459 |
0,929 |
0,636 |
0,800 |
0,601 |
|
6 |
da |
SVM |
28; 33; 42 |
0,782 |
0,800 |
0,790 |
0,579 |
0,929 |
0,727 |
0,840 |
0,678 |
|
7 |
dd |
LDA |
18; 34; 44 |
0,964 |
0,311 |
0,670 |
0,373 |
1,000 |
0,182 |
0,640 |
0,333 |
|
8 |
dd |
RF |
21; 31; 42 |
0,945 |
0,422 |
0,710 |
0,442 |
0,857 |
0,273 |
0,600 |
0,161 |
|
9 |
dd |
SVM |
12; 33; 36 |
0,618 |
0,578 |
0,600 |
0,195 |
1,000 |
0,091 |
0,600 |
0,230 |
|
10 |
vdw |
LDA |
9; 11; 12 |
0,855 |
0,689 |
0,780 |
0,554 |
0,786 |
0,273 |
0,560 |
0,068 |
|
11 |
vdw |
RF |
4; 6; 45 |
0,818 |
0,756 |
0,790 |
0,575 |
0,786 |
0,455 |
0,640 |
0,256 |
|
12 |
vdw |
SVM |
10; 45; 69 |
0,800 |
0,711 |
0,760 |
0,514 |
0,786 |
0,818 |
0,800 |
0,600 |
|
13 |
nn |
LDA |
19; 24; 38 |
0,909 |
0,600 |
0,770 |
0,543 |
0,929 |
0,364 |
0,680 |
0,363 |
|
14 |
nn |
RF |
24; 26; 30 |
0,836 |
0,733 |
0,790 |
0,574 |
0,929 |
0,545 |
0,760 |
0,524 |
|
15 |
nn |
SVM |
18; 38; 52 |
0,782 |
0,756 |
0,770 |
0,536 |
0,929 |
0,818 |
0,880 |
0,757 |
|
16 |
pp |
LDA |
7; 8; 11 |
0,855 |
0,578 |
0,730 |
0,454 |
0,929 |
0,364 |
0,680 |
0,363 |
|
17 |
pp |
RF |
10; 11; 66 |
0,818 |
0,667 |
0,750 |
0,492 |
0,786 |
0,545 |
0,680 |
0,342 |
|
18 |
pp |
SVM |
14; 17; 48 |
0,727 |
0,733 |
0,730 |
0,459 |
0,714 |
0,727 |
0,720 |
0,439 |
|
19 |
pn |
LDA |
6; 19; 72 |
0,900 |
0,600 |
0,770 |
0,543 |
0,786 |
0,364 |
0,600 |
0,165 |
|
20 |
pn |
RF |
8; 26; 42 |
0,836 |
0,756 |
0,800 |
0,595 |
0,786 |
0,545 |
0,680 |
0,342 |
|
21 |
pn |
SVM |
28; 30; 48 |
0,764 |
0,756 |
0,760 |
0,518 |
0,786 |
0,818 |
0,800 |
0,600 |
Примечание. Тип: a – акцептор, d – донор, vdw – ван-дер-ваальсовое взаимодействие, р – положительный заряд, n – отрицательный заряд. Номер дескриптора СМВВ соответствует расстоянию между атомами. Для получения расстояния нужно номер дескриптора умножить на 2 и разделить на 10. Например: 19 соответствует диапазону (19*2= 38, 38/10 = 3,8) = 3,8 – 3,6 ангстрема.
В отмеченных классификационных моделях используется 5 из 7 типов ААВ. Относительно низкая селективность этих типов свидетельствует о том, что информация, необходимая для конструирования QSAR моделей, достаточно равномерно распределена среди дескрипторов СМВВ. Что касается самих дескрипторов, то можно сделать вывод о том, что большая часть из них (11 из 15) попадают в интервал расстояний 5.4÷10.2 Å (рис. 2), что соответствует области несвязанных атомов.
Важным фактором, определяющим адекватность статистических QSAR зависимостей, является учет их AD [10]. Принимая это во внимание, на основе моделей 3, 6, 12, 15, 21 была проведена оценка их новых статистических параметров с использованием тестовой выборки (табл. 3).
Таблица 3
Статистические характеристики классификационных моделей (тестовая выборка) с учетом области применимости
|
№ |
n |
SN |
SP |
AC |
MCC |
|
6 |
24 |
0,929 |
0,700 |
0,833 |
0,657 |
|
12 |
24 |
0,786 |
0,800 |
0,792 |
0,580 |
|
21 |
24 |
0,786 |
0,800 |
0,792 |
0,580 |
В результате было установлено, что число соединений в тестовой выборке, которые соответствуют AD, уменьшается на единицу (с 25 до 24) в трех случаях из пяти (табл. 3). Это, очевидно, обусловлено случайным выбором молекул в тестовую выборку, что не гарантирует их попадания в AD.
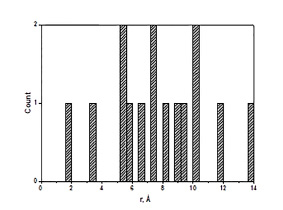
Рис. 2. Гистограмма межатомных расстояний для дескрипторов СМВВ
Таблица 4
Классификационные модели субстратной активности молекул по отношению к Р-гликопротеину, рассчитанные на основе HYBOT и фрактальных дескрипторов
|
Метод |
Дескрипторы |
Кросс-валидация (n = 100) |
Тестовая выборка (n = 25) |
||||||
|
SN |
SP |
AC |
MCC |
SN |
SP |
AC |
MCC |
||
|
LDA |
PSAed; Dval; Dunb* |
0,818 |
0,778 |
0,800 |
0,596 |
0,857 |
0,727 |
0,800 |
0,592 |
|
RF |
maxCa*maxCd; maxCa; PSAe |
0,800 |
0,756 |
0,780 |
0,556 |
0,857 |
0,818 |
0,840 |
0,675 |
|
SVM |
maxCa*maxCd; PSAe; Dval; Dunb |
0,764 |
0,800 |
0,780 |
0,561 |
0,857 |
0,727 |
0,800 |
0,592 |
Примечание. PSAed – ван-дер-ваальсова поверхность, пропорциональная донорным энтальпийным дескрипторам; Dval – валентный фрактальный дескриптор; Dunb* – фрактальная плотность несвязанных атомов; maxCa – максимальный акцепторный свободноэнергетический дескриптор; maxCd – максимальный донорный свободноэнергетический дескриптор; PSAe – ван-дер-ваальсова поверхность, пропорциональная сумме донорных и акцепторных энтальпийных дескрипторов; Dunb – несвязанный фрактальный дескриптор.
Интересно отметить, что это происходит за счет одного и того же соединения № 122. Наблюдаемое при этом снижение качества классификационных моделей является незначительным и связано, вероятно, в первую очередь с небольшим размером тестовой выборки.
В сравнительных целях были построены классификационные модели на базе дескрипторов компьютерной программы HYBOT [11] и ряда фрактальных дескрипторов [12]. Всего был рассчитан 41 дескриптор. После применения процедуры уменьшения пространства переменных, которая была использована по отношению к СМВВ дескрипторам, был получен ряд из 20 переменных. Методика построения моделей, а также обучающая и тестовая выборки были те же, что и для моделей на базе СМВВ. Результаты представлены в табл. 4.
Полученные QSAR модели имеют сопоставимые классификационные характеристики с соответствующими величинами моделей 3, 6, 12, 15 и 21. В качестве структурных параметров во всех HYBOT моделях фигурируют дескрипторы Н-связи. Важная роль водородной связи в процессах взаимодействия химических соединений с P-gp неоднократно упоминалась ранее [13]. Интересно отметить появление в классификационных моделях фрактальных дескрипторов. Эти независимые переменные являются новыми, и их влияние на связывание химических соединений с P-gp требует дальнейшего изучения. При этом следует подчеркнуть, что HYBOT дескрипторы являются двумерными интегральными характеристиками. Это ограничивает их использование при анализе и генерации трехмерных структур в отличие от дескрипторов на основе СМВВ.
Заключение
Таким образом, с использованием методов линейного дискриминантного анализа, случайного леса, опорных векторов и дескрипторов на базе спектров межатомных внутримолекулярных взаимодействий сконструированы классификационные модели, позволяющие априори отделять соединения с субстратной активностью по отношению к Р–гликопротеину от соединений, не обладающих такой активностью. Лучшие две модели, обладающие удовлетворительными статистическими характеристиками и ясной физико-химической интерпретацией, могут быть рекомендованы к практическому использованию для непосредственного отнесения веществ к одному из классов субстрат/несубстрат к Р–gp, а также для анализа пространственной структуры молекул и генерирования гипотез при поиске новых субстратов к P–gp. Проведено сравнение классификационных моделей, построенных на базе дескрипторов СМВВ, и моделей на базе дескрипторов, рассчитанных с помощью компьютерной программы HYBOT. Показаны преимущества моделей на базе дескрипторов СМВВ.
Библиографическая ссылка
Раздольский А.Н., Григорьев В.Ю., Ярков А.В., Григорьева Л.Д., Страхова Н.Н., Казаченко В.П., Раевская, О.Е., Раевский О.А. КЛАССИФИКАЦИОННЫЕ QSAR МОДЕЛИ СУБСТРАТНОЙ АКТИВНОСТИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ ПО ОТНОШЕНИЮ К P-ГЛИКОПРОТЕИНУ НА БАЗЕ СПЕКТРА МЕЖАТОМНЫХ ВНУТРИМОЛЕКУЛЯРНЫХ ВЗАИМОДЕЙСТВИЙ // Международный журнал прикладных и фундаментальных исследований. 2021. № 6. С. 97-103;URL: https://applied-research.ru/en/article/view?id=13238 (дата обращения: 20.07.2026).
DOI: https://doi.org/10.17513/mjpfi.13238