
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
CHAPERONE HSP70 MODULES THE PATHWAY OF RAW264.7 CELL ENDOCYTOSIS OF AT LPS-INDUCED ACTIVATION

Эндотоксины (липополисахариды (LPS)) – термостабильные компоненты наружной части клеточной мембраны грамотрицательных микроорганизмов микробиоты человека. Поступая в кровь из кишечника (при эндотоксемии), эндотоксины участвуют в патогенезе грамотрицательного сепсиса, метаболического синдрома, диабета типа 2, почечной недостаточности, сердечно-сосудистых, нейродегенеративных и других заболеваний [1, 2]. Основными клетками-мишенями эндотоксинов являются циркулирующие в кровяном русле нейтрофилы и моноциты, а также макрофаги, дифференцирующиеся из моноцитов в различных тканях организма человека и животных. Макрофаги в организме при воспалительных процессах выполняют различные функции, такие как презентация антигена, фагоцитоз, синтез и секреция различных медиаторов воспаления. Активация клеток-мишеней липополисахаридами происходит при связывании LPS с TLR4 рецептором. Этой активации предшествует последовательность реакций, которые начинаются LPS-связывающим белком (LBP), извлекающим молекулу LPS из липополисахаридных мицелл, находящихся в кровяном русле. Далее LBP доставляет молекулу LPS к мембранному гликопротеину CD14 макрофагов, который передает LPS MD2 белку. Затем образуются два комплекса, состоящие из LPS, MD2 и TLR4. После их агрегации образуется гетеродимерный рецепторный комплекс [MD2-LPS-TLR4]2. Для активации клеток регуляторные сигналы от упомянутого рецепторного комплекса через различные пути трансдукции сигналов передаются к ядерным факторам транскрипции клеток [2]. После активации под действием LPS макрофаги секретируют провоспалительные цитокины (IL-1, 1β, 2, 6, 8), фактор некроза опухолей TNFα, генерируют оксид азота (NO) и активные формы кислорода (АФК). Под действием LPS происходит увеличение экспрессии ряда клеточных генов, включая индуцибельную синтазу оксида азота (iNOS), TNFα, рецепторов LPS (TLR4) и других белков [3]. В предыдущих наших работах было опубликовано, что экзогенный Hsp70 защищает разные типы клеток от их активации LPS, уменьшая LPS-индуцированную продукцию АФК, NO, TNFα [4, 5]. В настоящее время известны различные пути эндоцитоза в клетки [6]. Известно, что LPS поступают в клетки-мишени с помощью рецептор-зависимого эндоцитоза [2], а Hsp70 – с помощью клатрин-независимого эндоцитоза [7]. В связи с этим представляет интерес исследовать действие шаперона Hsp70 на различные пути эндоцитоза LPS в клетки.
Цель исследования – определить отдельные пути эндоцитоза шаперона Hsp70 и сравнить их с путями эндоцитоза LPS в клетки RAW264.7.
Материалы и методы исследования
Культуры клеток. Клетки мышиных макрофагов RAW264.7 были получены из American type culture collection. Клетки L-929 (мышиные фибробласты) получены из Российской коллекции клеточных культур (ИНЦ РАН). Клетки RAW264.7 и L-929 выращивали в культуральной среде (КС) DMEM (Sigma-Aldrich, США), содержащей 10 % термоинактивированной эмбриональной телячьей сыворотки, которая была проверена на присутствие эндотоксина (0,01 ед/мл) («HyClone», США), содержала 2 мМ L-глутамина и стандартный набор антибиотиков. Клетки выращивали в СО2-инкубаторе при 37 °C в атмосфере, содержащей 5 % CO2.
Получение Hsp70. Использовали рекомбинантный человеческий Hsp70 (очищенный от LPS), экспрессированный в клетках Spodoptera frugiperda согласно [4].
Продукцию фактора некроза опухолей TNFα клетками RAW264.7 определяли по цитотоксическому действию образцов на клетки-мишени [5].
Продукцию оксида азота NO клетками измеряли с использованием реактива Грисса [4] (Sigma-Aldrich, США).
Продукцию активных форм кислорода (АФК) клетками RAW264.7 измеряли с использованием красителя нитросинего тетразолия НСТ [5].
Эксперименты c использованием ингибиторов. Ингибиторы эндоцитоза добавляли к клеткам за 30 мин до добавления белка Hsp70. В работе были использованы следующие ингибиторы: динасор – ингибитор клатрин-опосредованного эндоцитоза, амилорид – ингибитор макропиноцитоза, филипин – ингибитор кавеолин-зависимого эндоцитоза, нокодазол – ингибитор тубулинзависимого эндоцитоза, метил-бета-циклодекстрин – ингибитор липидных микродоменов. В ходе предварительных экспериментов и полученной концентрационной зависимости были определены оптимальные и нетоксичные концентрации используемых ингибиторов (данные не представлены).
Клетки RAW264.7 собирали с использованием скреперов фирмы Sarstedt (Германия), отмывали в полной КС, подсчитывали и разводили КС, содержащей 10 % ЭТС, до требуемой концентрации, рассеивали в 24-луночные планшеты и культивировали в течение 24 ч при 37 °C и 5 % CO2. Заменяли КС на свежую, добавляли ингибиторы эндоцитоза на 30 мин, делали добавки Hsp70 на 2 ч, затем добавляли LPS и инкубировали клетки в течение 24 ч при 37 °C и 5 % CO2. Выбранная схема проведения экспериментов позволяла определять продукцию активных форм кислорода, оксида азота и фактора некроза опухолей в одних и тех же опытных пробах.
Статистика
Все эксперименты с клетками проводились в четырех повторностях. Результаты анализировали с помощью программы SigmaPlot. Различия между группами были проанализированы с помощью дисперсионного анализа ANOVA (Shapiro-Wilk’s test, p < 0,05). Данные представлены как среднее значение ± стандартное отклонение в четырех независимых экспериментах, выполненных в виде четырехкратных повторностей.
Результаты исследования и их обсуждение
Полученные результаты (рис. 1–3) показали, что все использованные ингибиторы снижали продукцию АФК, NO и TNFα, активированную LPS. Hsp70 защищал клетки от действия LPS (рис. 1–3, столбики CM, 2), уменьшая продукцию АФК, NO и TNFα (рис. 1–3, столбики CM, 3). Для сравнительного анализа путей эндоцитоза LPS с путями эндоцитоза шаперона Hsp70 в присутствии ингибиторов эндоцитоза мы рассчитывали отношение продукции АФК, NO, TNFα в пробах с LPS (рис. 1–3, столбики CM, 2) (n1) к образованию измеряемых соединений в образцах с (Hsp70 + LPS) (рис. 1–3, столбики CM, 3) (n2). Для продукции АФК (рис. 1) эти величины (n1/n2) равны, соответственно, 2 и 1.2. Исходя из этих расчетов и данных рисунков видно, что ингибиторы амилорид, динасор, MbCD и нокодазол практически полностью отменяли защиту клеток Hsp70 от образования АФК под действием LPS (рис. 1), сравнение столбиков 2 и 3. Филипин несколько снижал защитное действие Hsp70 (рис. 1, Fil, 3). Действие динасора на продукцию NO (рис. 2) было подобно ингибированию продукции АФК (рис. 1, Dyn, 3). Динасор продемонстрировал практически полную отмену защитного действия Hsp70 при регистрации продукции оксида азота клетками (рис. 2). Все остальные ингибиторы отменяли защиту Hsp70 не полностью. Менее эффективным был амилорид (рис. 2). Действие ингибиторов эндоцитоза на продукцию клетками TNFα, показало, что амилорид, динасор и нокодазол блокировали защитное действие Hsp70.
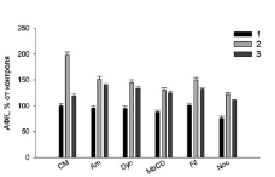
Рис. 1. Действие ингибиторов эндоцитоза на генерацию АФК в клетках RAW264.7 в присутствии Hsp70 (1 мкг/мл) и LPS (1 мкг/мл). CМ – культуральная среда; Am – 100 мкМ амилорид; Dyn – 40 мкМ динасор; MbCD – 2 мМ метил-β-циклодекстрин; Fil – 1 мкМ филипин; Noc – 1 мкМ нокодазол. 1 – CM; 2 – добавление LPS; 3 – добавление Hsp70 и через 2 ч LPS. Уровень АФК в контроле равен 100 %, остальные величины – проценты от контроля
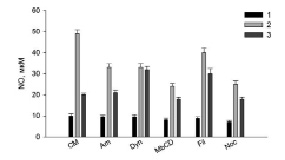
Рис. 2. Продукция оксида азота (NO) клетками RAW264.7 при действии ингибиторов эндоцитоза в присутствии Hsp70 и LPS. Обозначения и условия эксперимента как на рис. 1
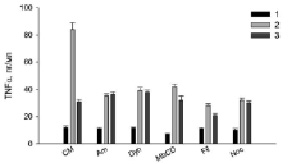
Рис. 3. Влияние ингибиторов эндоцитоза на продукцию TNFα клетками RAW264.7 в присутствии Hsp70 и LPS. Обозначения и условия эксперимента как на рис. 1
При активации липополисахаридами TLR4 рецептора макрофагов продуцируемый клетками TNFα может связываться с рецепторами TNFα этих клеток. В результате этого в клетках активируется сборка NOX2 и, как следствие, происходит значительное увеличение продукции АФК клетками [8]. В функционировании НАДФН-оксидазы важную роль играют липидные микродомены, разрушение которых MbCD снижает продукцию АФК клетками [9], как и показывают наши экспериментальные данные (рис. 1, MbCD, 2, 3). В липидных микродоменах функционируют рецепторы TNFα и LPS [10]. Ингибитор липидных микродоменов – MbCD снижает уровень секреции TNFα и NO макрофагами (рис. 2 и 3, MbCD, столбики 2 и 3). Поэтому снижение защиты клеток от LPS белком Hsp70, по-видимому, связано с разрушением липидных микродоменов MbCD и, как следствие уменьшением проникновения Hsp70 в клетки и снижением эффективности его защиты. Полученные нами данные по действию MbCD подтверждаются результатами о механизмах внутриклеточной регуляции воспалительного ответа клеток на LPS Установлено, что активация TLR4 рецепторов запускает два сигнальных пути: MyD88-зависимого и TRIF-зависимого. Эти сигнальные пути приводят к производству провоспалительных цитокинов. MyD88- и TRIF-зависимые сигнальные пути запускаются последовательно и связаны с перераспределением LPS-активированного TLR4 из плазматической мембраны в эндосомы. Транслокация TLR4 внутрь клетки происходит в эндосомах, и его дальнейшая лизосомная деградация способствует прекращению воспалительного ответа [2]. LPS-индуцированные MyD88-зависимые сигнальные пути оказывают модулирующее действие на клеточный метаболизм. Было показано, что TRAF6 активирует киназу Akt, приводя к быстрому усилению гликолиза. Гликолиз, стимулированный LPS, и последующий синтез ацетил-КоА жирных кислот de novo может способствовать ацетилированию гистонов, необходимому для транскрипции и расширения эндоплазматической сети и аппарата Гольджи и также необходим для интенсивной выработки и секреции цитокинов [2].
Различные белки поступают в клетки преимущественно за счет клатринзависимого эндоцитоза [11]. В наших экспериментах динасор – ингибитор клатринзависимого эндоцитоза – существенно снижал активированную LPS продукцию АФК, NO и TNFα клетками RAW264.7, а также снижал защитное действие Hsp70 (рис. 1–3, столбики 3). Обнаруженное в экспериментах уменьшение продукции АФК, NO и TNFα клетками связано со снижением встраивания рецепторов CD14 и TLR4 в мембрану RAW264.7 [11]. Уменьшение генерации и TNFα в присутствии данного ингибитора (рис. 1, 3) обусловлено ингибированием NADPH-оксидазы [12]. Снижение защитного действия Hsp70 динасором связано с уменьшением содержания в клеточной мембране TLR4 рецептора.
Известно, что амилорид является модулятором протон-активируемых каналов клетках, он блокирует продукцию провоспалительных цитокинов, индуцированную LPS [13]. В наших экспериментах амилорид эффективно снижал LPS-активированную продукцию АФК, NO и TNFα (рис. 1–3): величина n1/n2 составила 1,09; 1,57 и 1,24 для АФК, NO и TNFα, соответственно. Амилорид ингибирует активность Na+/H+-обменника и вызывает снижение LPS-индуцированной активации НАДФН-оксидазы. Ингибирование Na+/H+-обменника снижает продукцию TNFα макрофагами. Hsp70 может связываться с Na+/H+-обменником . С этим связано снижение защиты белком клеток RAW264.7 от действия LPS [13].
Тубулины играют важную роль в воспалительном ответе макрофагов на липополисахариды, в частности регулируют LPS-индуцированную активацию макрофагов через сигнальный путь MAPK/p38 [14]. Пути внутриклеточной сигнализации с участием митогенактивированных киназ и p38MAPK участвуют в регуляции продукции АФК, NO и TNFα [2]. В наших экспериментах нокодазол (ингибитор тубулин-зависимого эндоцитоза) снижал образование АФК, NO, TNFα клетками. Снижение нокодазолом защитного действия Hsp70 обусловлено, по-видимому, воздействием этого ингибитора эндоцитоза на TLR4-зависимые пути внутриклеточной сигнализации клеток RAW264.7 [14].
Кавеолинзависимый эндоцитоз занимает важное место среди других механизмов эндоцитоза [15]. Кавеолы небольшие колбообразные впячивания плазматической мембраны участвуют во множестве клеточных процессов, среди которых – мембранный транспорт и формирование клеточного ответа на внешний сигнал. В них содержится белок – кавеолин, липиды (холестерин и сфинголипиды), мембранные рецепторы, синтаза оксида азота Филипин – ингибитор кавеол-зависимого эндоцитоза – в наших экспериментах снижал величину n1/n2, которая составила для генерации АФК-1,14 и 1,33 для секреции оксида азота и TNFα.
Заключение
Полученные нами результаты позволяют сделать вывод о том, что человеческий рекомбинантный белок Hsp70 снижает LPS-активированную продукцию АФК, NO, TNFα. Проведенный ингибиторный анализ показал, что защита клеток RAW264.7 LPS-активированной продукции АФК, NO, TNFα экзогенным белком Hsp70 осуществляется с участием кавеолин-, тубулин-, клатрин- и рецепторзависимого эндоцитоза, иакже в этом механизме участвует пиноцитоз.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект № 19-04-00109).
Библиографическая ссылка
Юринская М.М., Евгеньев М.Б., Афанасьев В.Н., Винокуров М.Г. ШАПЕРОН HSP70 МОДУЛИРУЕТ ПУТИ ЭНДОЦИТОЗА КЛЕТОК RAW264.7 ПРИ LPS-ИНДУЦИРОВАННОЙ АКТИВАЦИИ // Международный журнал прикладных и фундаментальных исследований. 2021. № 12. С. 22-26;URL: https://applied-research.ru/en/article/view?id=13324 (дата обращения: 10.08.2026).
DOI: https://doi.org/10.17513/mjpfi.13324