
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
CELL DEATH AND FUNCTIONAL ACTIVITY OF NEUTROPHILS IN PATIENTS WITH CHRONIC HEART FAILURE WITH ENDOTOXEMIA

Хроническая сердечная недостаточность (ХСН) является одной из самых важных проблем современной медицины [1]. В России распространенность ХСН I–IV функционального класса (ФК), по классификации New York Heart Association Functional (NYHA), составляет 7–10 % случаев, а абсолютное число больных ХСН по состоянию на 2018 г. равняется 12,35 млн чел., годовая смертность пациентов с сердечной недостаточностью составляет в России примерно 12 %. В патогенезе сердечной недостаточности критическую роль играют воспалительные процессы. Значительным инициатором воспаления являются эндотоксины (липополисахариды (LPS)). При сердечной недостаточности довольно часто наблюдается эндотоксемия, которая возникает вследствие поступления молекул LPS из бактерий микробиома кишечника человека в кровяное русло [2]. Работы последнего времени показали, что состав кишечной микробиоты связан с воспалением, ожирением и с нарушением обмена веществ. Установлено, что диета с большим количеством жиров и углеводов вызывает метаболическую эндотоксемию в крови больных с этими патологиями [3].
Поступающие в кровь LPS, связываясь с мембранными рецепторами TLR4, праймируют нейтрофилы, после чего активационный сигнал от рецептора передается к различным внутриклеточным сигнальным путям, что приводит к продуцированию провоспалительных цитокинов, активных форм кислорода (АФК), оксида азота, фактора некроза опухолей (TNFα) [4]. Гибель кардиомиоцитов играет ключевую роль при СН. Выявлено, что в механизмах активации апоптоза кардиомиоцитов принимают участие рецептор липополисахаридов TLR4 и NADPH-оксидаза 4 сердца [5].
В физиологических условиях нейтрофилы находятся в кровотоке не более одних суток, затем поступают в ткани, где могут дополнительно функционировать 1–2 дня до активации апоптотической гибели. Далее макрофаги осуществляют эффероцитоз нейтрофилов, что обеспечивает безвоспалительное удаление этих клеток из организма [6].
LPS вызывает ингибирование апоптоза нейтрофилов при бактериальной инфекции (грамотрицательный сепсис), сахарном диабете, сердечно-сосудистых, нейродегенеративных заболеваниях [7]. Под действием LPS в нейтрофилах увеличивается продукция АФК, провоспалительных цитокинов и различных медиаторов воспаления [8]. Следует отметить, что поступающие в кровь липополисахариды воздействуют не только на нейтрофилы. LPS увеличивают продукцию АФК и провоспалительных цитокинов моноцитов, макрофагов и клеток эндотелия. LPS ускоряют апоптоз этих клеток.
Помимо апоптоза и некроза нейтрофилы могут погибать посредством нетоза – программируемой клеточной гибели, сопровождаемой выбросом из погибающих нейтрофилов экстраклеточных ловушек нейтрофилов Neutrophil extracellular traps (NET), содержащих фрагменты ДНК, ядерные белки, миелопероксидазу и эластазу. Нейтрофильные экстраклеточные ловушки захватывают и убивают бактериальные клетки [9]. Однако когда воспаление продолжается или сохраняется возбудитель, нейтрофилы, высвобождая NET, усугубляют повреждение тканей и усиливают воспалительные процессы в организме [10]. Недавние исследования показывают, что нетоз участвует не только в инфекциях, но и в других заболеваниях. Совсем недавно было показано, что образование NET способствует венозной тромбоэмболии, а также и прогрессированию атеросклероза, вызывая венозный и артериальный тромбозы. В NET попадают тромбоциты, эритроциты, моноциты, что, в свою очередь, вызывает образование больших тромбов в сосудистом русле. Обнаружено присутствие NET в просвете атеросклеротических сосудов человека и препаратах коронарных артерий, полученных от больных после острого инфаркта миокарда [11].
Целью исследования явилось изучение клеточной гибели (апоптоза и нетоза) и функциональной активности нейтрофилов у больных хронической сердечной недостаточностью при эндотоксемии.
Материалы и методы исследования
В процессе проведения исследования были сформированы группы наблюдения по критериям включения и данным клинического и биохимического анализов крови, проведена верификация диагноза, набрано 20 пациентов с диагнозом хроническая сердечная недостаточность. Критериями включения являлись: возраст больных ≥ 30 лет, III функциональный класс хронической сердечной недостаточности по классификации NYHA, и фракция выброса ≤ 40 %. У включенных в исследование больных уровень сывороточного мозгового натрийуретического пептида (BNP) составлял ≥ 150 пг/мл или N-концевого про-BNP (NT-proBNP) ≥ 600 пг/мл, если пациенты были госпитализированы в стационар по поводу сердечной недостаточности в течение года до включения в исследование, то уровень BNP составлял ≥ 100 пг/мл, а NT-proBNP ≥ 400 пг/мл. Для выявления эндотоксемии у больных с ХСН определяли уровень LPS в плазме крови с использованием LAL-теста (0,25±0,02 ед/мл (контрольная группа – менее 0,16 ед/мл)). Средний возраст больных ХСН 64,2 ± 8,0, женщины – 9 чел, мужчины – 11 чел., количество пациентов, перенесших инфаркт миокарда, – 3 чел., количество пациентов, перенесших операцию на сердце, – 6 чел. Также были обследованы здоровые индивидуумы (n = 19) обоих полов (доноры-добровольцы), средний возраст 53,6 ± 9,9.
Нейтрофилы выделяли из периферической крови больных, участвующих в исследовании, с помощью метода дифференциального центрифугирования на двухслойном градиенте плотности перколла (70 % и 55 %). Осаждённые клетки ресуспендировали в фосфатно-солевом буфере или в культуральной среде DMEM, содержащей 10 % термоинактивированной эмбриональной сыворотки теленка, тестированной на присутствие эндотоксинов, 2 мМ L-глутамина, стандартный набор антибиотиков (все реактивы фирмы Sigma-Aldrich, США).
Генерацию активных форм кислорода (АФК) в клетках определяли с использованием красителя нитросинего тетразолия (НСТ) [12]. Продукцию АФК нейтрофилами контрольной группы принимали за 100 %.
Регистрацию гибели клеток (апоптоз, некроз и нетоз) проводили методом флуоресцентной микроскопии (инвертированный микроскоп Keyence, Япония). Для регистрации апоптоза и некроза использовали флуоресцентные зонды Hoechst 33342 и Propidium iodide (PI). Выделенные нейтрофилы (106 клеток/мл) культивировали в CO2 инкубаторе в течение 15 ч (37 °С, 5 % CO2) в полной культуральной среде. Далее добавляли на 30 мин к клеткам 1 мкг/мл Hoechst 33342 и за 5 мин до регистрации 30 мкМ PI. Для регистрации нетоза использовали флуоресцентный зонд Sytox Green (Invitrogen, США). Нейтрофилы инкубировали 3 ч в тех же условиях, что и для обнаружения апоптоза. Затем к клеткам на 30 мин добавляли 0,8 мкМ Sytox Green. Регистрировали нетоз, активированный 20 нM форбол-12-миристат-13-ацетатом (PMA), и спонтанный нетоз (без PMA).
Уровни TNFα и интерлейкинов IL-1β и IL-6 были определены с использованием метода иммуноферментного анализа ELISA. Бесклеточные супернатанты собирали для определения концентрации цитокинов с использованием наборов: набор реагентов для иммуноферментного определения концентрации фактора некроза опухолей – альфа «Альфа-ФНО-ИФА-БЕСТ», набор реагентов для иммуноферментного определения концентрации человеческого интерлейкина-1 бета «Интерлейкин-1 бета- ИФА-БЕСТ», набор реагентов для иммуноферментного определения концентрации человеческого интерлейкина-6 «Интерлейкин-6- ИФА-БЕСТ», («ВЕКТОР-БЕСТ», Россия), в соответствии с протоколами производителя.
Уровень эндотоксинов в крови больных определяли с использованием LAL-теста по протоколу фирмы-производителя Sigma-Aldrich, США.
Использование ингибиторов
В работе исследовали действие следующих ингибиторов: TAK242 – ингибитор TLR4-зависимого сигнального пути, SB203580 – ингибитор р38MAPK (р38 митоген-активируемая протеинкиназа), дифенилениодоний хлорида (DPI) – ингибитор НАДФН-оксидазы, вортманнин (Wortmannin) – ингибитор фосфоинозитол-3 киназы (PI3K) и SP600125 – ингибитор янус киназы (JNK). Рабочие концентрации ингибиторов подбирали по их минимальной токсичности. Ингибиторы добавляли к клеткам на 30 мин, после чего клетки отмывали от ингибиторов и использовали для дальнейшей работы.
Уровень TLR4 в клеточной мембране нейтрофилов определяли, используя антитела Anti-TLR4 (Toll-Like Receptor 4, CD284), ковалентно меченые флуоресцентным зондом фикоэритрином (PE) (USBiological, T8050-34D). После выделения нейтрофилов их окрашивали Anti-TLR4 антителами согласно рекомендациям производителя при температуре 0 °C в течение 30 мин. Определение уровня TLR4 в нейтрофилах выполняли на проточном цитометре CytoFlex S (Beckman Coulter, США) (сразу после завершения окрашивания), регистрируя Median Fluorescent Intensity (MFI), окрашенных антителами нейтрофилов.
Статистическая обработка данных
Экспериментальные результаты анализировали в SigmaPlot. Различия между экспериментальными группами определяли дисперсионным анализом ANOVA (Shapiro-Wilk’s test, p < 0,05).
Результаты исследования и их обсуждение
Основными повреждающими ткани организма факторами являются АФК. Как известно, нейтрофилы, как и все фагоциты, при активации LPS продуцируют активные формы кислорода (АФК). Основной вклад в пул АФК, производимый нейтрофилами, вносит супероксид анион радикал, генерируемый НАДФН-оксидазой этих клеток. Активация НАДФН-оксидазы при действии LPS происходит вследствие активации различных сигнальных путей передачи сигнала от TLR4. Для поиска новых подходов к лечению сердечно-сосудистых патологий необходимо понимать механизмы, лежащие в основе увеличения уровня провоспалительных цитокинов при этих заболеваниях. Поэтому в работе были изучены различные сигнальные внутриклеточные пути, активацию которых вызывают липополисахариды.
В данном исследовании мы сравнили роль отдельных сигнальных путей нейтрофилов пациентов с ХСН (рис. 1, Б) с нейтрофилами здоровых доноров (рис. 1, А). Для этой цели клетки обрабатывали специфическими ингибиторами этих сигнальных путей. Ингибитор DPI использовали для подтверждения специфичности регистрируемых данных.
Полученные результаты показали, что все ингибиторы, за исключением Wortmannin (рис. 1, А, столбики 1–3, 5), немного снижают продукцию АФК нейтрофилами контрольной группы (рис. 1, А, столбик К). В группе больных с ХСН продукция АФК в нейтрофилах в пробах без ингибиторов была значительно выше, чем в нейтрофилах контрольной группы (рис.1, Б, К по сравнению с рис.1, А, К). При этом все ингибиторы снижали продукцию АФК нейтрофилами (рис. 1, Б, столбики 1–5) по сравнению с нейтрофилами без воздействия ингибиторов (рис. 1, Б, столбик К). Причем ингибирование всеми ингибиторами было значительно больше, чем для нейтрофилов контрольной группы (рис. 1).
Таким образом, полученные результаты показали, что в регуляции продукции АФК нейтрофилами больных с ХСН с эндотоксемией важную роль играют TLR4-, p38MAPK-, PI3K- и JNK-зависимые сигнальные пути. Использованные ингибиторы более эффективно снижают продукцию АФК нейтрофилами пациентов с ХСН по сравнению с нейтрофилами контрольной группы лиц.
Действие LPS, помимо продуцирования АФК, вызывает синтез различных провоспалительных цитокинов [5, 7]. Наши исследования функциональных свойств нейтрофилов больных с ХСН показали, что в данном случае наблюдается увеличение продукции провоспалительных цитокинов (по сравнению с нейтрофилами контрольной группы): TNFα, IL-1b, IL-6 (рис. 2, столбики 2 по сравнению со столбиками 1).
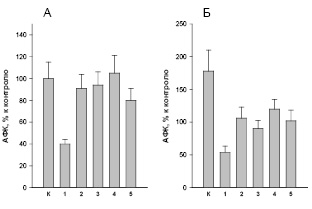
Рис. 1. Влияние ингибиторов сигнальных путей на продукцию АФК нейтрофилами больных с ХСН. А – контрольная группа пациентов, Б – больные с ХСН. Продукция АФК нейтрофилами контрольной группы (в отсутствии ингибиторов) принята за 100 %. Ингибиторы: 1 – 10 мкМ diphenyleneiodonium chloride (DPI); 2 – 1 мкг/мл TAK242; 3 – 1 мкМ SB203580; 4 – 50 нМ Wortmannin (Wort); 5 – 2 мкМ SP600125
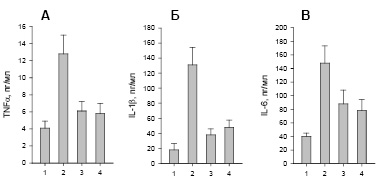
Рис. 2. Влияние ингибиторов сигнальных путей на продукцию TNFα, IL-1β, IL-6 нейтрофилами больных с ХСН. 1 – контрольная группа, 2–4 – группа больных с ХСН; 3 – воздействие ингибитора TAK242 (1 мкг/мл), 4 – воздействие ингибитора 1 мкМ SB203580
Изучение влияния ингибиторов на продукцию АФК нейтрофилами показало, что наибольшим ингибирующим эффектом обладал ингибитор p38MAPK (рис. 1, Б, 3). Поэтому далее было изучено действие SB203580 и TAK242 (блокирующего передачу всех сигналов от TLR4) на продукцию TNFα, IL-1b, IL-6. Полученные результаты показали, что TAK242 значительно снижает продукцию TNFα, IL-1β (рис. 2, А и Б, столбики 3). Воздействие TAK242 на продукцию IL-6 несколько слабее (рис. 2, В, столбик 3). Ингибитор SB203580 также значительно сильнее ингибировал продукцию TNFα, IL-1β (рис. 2, А и Б, столбики 4) по сравнению с воздействием на продукцию IL-6 (рис. 2, В, столбик 4).
Таким образом, полученные результаты показали, что в регуляции продукции TNFα, IL-1b, IL-6 нейтрофилами больных с ХСН с эндотоксемией важную роль играют TLR4- и p38MAPK-зависимые сигнальные пути.
В последние годы было показано, что АФК, продуцируемые НАДФН-оксидазой, участвуют в качестве сигнальных молекул в активации апоптоза и нетоза нейтрофилов [13]. Сравнительный анализ клеточной гибели нейтрофилов пациентов с ХСН и нейтрофилов лиц из контрольной группы показал, что у пациентов с ХСН значительно ингибирован апоптоз нейтрофилов и увеличен спонтанный и активированный PMA нетоз (таблица).
Гибель нейтрофилов у здоровых лиц и у больных ХСН
|
Гибель нейтрофилов |
Здоровые |
Больные ХСН |
|
Апоптоз, % |
100±8 |
78±6 |
|
Спонтанный нетоз, % |
1,5±0,8 |
5,9±1,5 |
|
Активированный нетоз, % |
14,6±3,8 |
28,9±6,8 |
Полученные результаты по регистрации генерации АФК, TNFα, IL-1b, IL-6 нейтрофилами с использованием ингибиторов сигнальных путей показали, что в процессах увеличения уровня этих медиаторов воспаления ключевую роль играют TLR4-зависимые и p38MAPK-зависимые сигнальные пути. Последующий анализ уровня TLR4 в клеточной мембране нейтрофилов, регистрируемый по уровню медианной интенсивности флуоресценции антител, показал, что изменение клеточной гибели и функциональной активности нейтрофилов при эндотоксемии в значительной степени обусловлено увеличением уровня TLR4 в мембране нейтрофилов больных ХСН (2021 ± 140) (по сравнению с нейтрофилами контрольной группы- 1582 ± 140).
Заключение
Полученные нами результаты по сравнению функциональных показателей контрольных нейтрофилов и нейтрофилов больных ХСН показали, что при эндотоксемии значительно увеличивается уровень TNFα, IL-1β, IL-6, АФК, уровень рецепторов TLR4, значительно снижается апоптоз нейтрофилов и увеличивается их нетоз, что имеет большое значение в патогенезе хронической сердечной недостаточности.
Работа выполнялась в рамках Госзадания по теме 0576-2020-0005, одобрена ЛЭК БПНЦ РАН, протокол № 14 от 04.09.2018 г.
Библиографическая ссылка
Юринская М.М., Винокуров М.Г., Сусликов А.В. КЛЕТОЧНАЯ ГИБЕЛЬ И ФУНКЦИОНАЛЬНАЯ АКТИВНОСТЬ НЕЙТРОФИЛОВ У БОЛЬНЫХ ХРОНИЧЕСКОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТЬЮ ПРИ ЭНДОТОКСЕМИИ // Международный журнал прикладных и фундаментальных исследований. 2022. № 8. С. 15-19;URL: https://applied-research.ru/en/article/view?id=13420 (дата обращения: 26.07.2026).
DOI: https://doi.org/10.17513/mjpfi.13420