
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
DRENOBLOCKERS PROPRANOLOL AND FENTOLAMINE MODULATE LPS-INDUCED INTRACELLULAR SIGNALING OF HUMAN NEUTROPHILS

Хроническая сердечная недостаточность (ХСН) является актуальной проблемой здравоохранения и одной из основных причин смертности во всем мире. Исследования последнего десятилетия показали, что патофизиология сердечной недостаточности (CH) в значительной степени связана с нарушением перфузии кишечника, что приводит к дисфункции кишечного эндотелиального барьера, который поддерживается несколькими механизмами хорошо сбалансированной кишечной микробиоты [1]. У пациентов с СН происходит снижение кровотока в кишечном эндотелии из-за снижения сердечного выброса. Это, в свою очередь, приводит к ишемии кишечной стенки и увеличению проницаемости из-за структурного нарушения барьерной функции кишечного эпителия [2]. Кроме того, системный застой у пациентов с СН может вызвать отек кишечной стенки, что также приводит к повышению кишечной проницаемости, которая, в свою очередь, увеличивает транслокацию эндотоксинов (липополисахаридов (LPS)) и других компонентов, продуцируемых грамотрицательными бактериями, в системный кровоток, вызывая эндотоксемию [3]. Этот процесс может дополнительно активировать синтез цитокинов и вызвать системное воспаление, которое, в свою очередь, способствует прогрессированию сердечной недостаточности [4].
Действие LPS реализуется благодаря взаимодействию с Толл-подобными рецепторами 4 (Toll-like receptor 4, TLR4) в клетках врожденного иммунитета (в том числе и нейтрофилах), клетках эндотелия, а также в кардиомиоцитах. В ответ на действие LPS клетки различных тканей организма продуцируют активные формы кислорода, цитокины. Под действием LPS происходит ингибирование апоптоза нейтрофилов [5]. Нейтрофилы традиционно рассматриваются как терминально дифференцированные короткоживущие фагоциты с довольно неконтролируемым образом действий. Однако в последнее время было выяснено, что эти клетки могут выступать в качестве регуляторов воспалительных процессов в сердечно-сосудистой системе. Показано, что после ишемического инсульта нейтрофилы усиливают дисфункцию клеток эндотелия за счет секреции нейтрофильной эластазы и активных форм кислорода (АФК). Нейтрофильные внеклеточные ловушки (Neutrophil Extracellular Traps (NET)) способствуют росту тромбов, NET ускоряют гибель нейронов. В то же время установлено, что в местах повреждения артерий активированные нейтрофилы посредством пептида кателицидина, сосудистого эндотелиального фактора роста и эпидермального фактора роста способствуют восстановлению клеток эндотелия. Нейтрофилы также влияют на заживление сердца после инфаркта миокарда, стимулируя переход макрофагов в сторону репаративного фенотипа (который активно фагоцитирует поврежденные клетки). Кроме этого, нейтрофилы, циркулирующие в сердце, секретируют аннексин А1, который способствует дифференцировке макрофагов в макрофаги проангиогенного фенотипа, что, в свою очередь, способствует ангиогенезу в ишемизированном сердце [6].
При сердечно-сосудистых патологиях важную роль играют адренорецепторы. В лечебной практике достаточно широко используются неселективные блокаторы адренорецепторов – пропранолол (блокирует β1- и β2-адренорецепторы) и фентоламин (блокирует α1- и α2-адренорецепторы). Пропранолол эффективно применяется при лечении СН у детей [7]. Кроме того, пропранолол снижает LPS- индуцированную секрецию TNFα моноцитами человека [8]; снижает экспрессию матриксной металлопротеазы 9 (играет важную роль в патогенезе атеросклероза), экспрессия которой увеличивается при действии норадреналина и LPS [9]. Фентоламин, как и пропранолол, эффективно используется при лечении СН в педиатрии [10]. Однако механизмы действия пропранолола и фентоламина на генерацию АФК и апоптоз нейтрофилов при действии LPS остаются недостаточно исследованными.
Целью исследования явилось изучение влияния адреноблокаторов фентоламина и пропранолола на апоптоз и продукцию АФК нейтрофилами больных хронической сердечной недостаточностью при действии липополисахаридов.
Материалы и методы исследования
Нейтрофилы выделяли из крови больных, которые принимали участие в исследовании, в которое были включены согласно критериям включения (возраст ≥35 лет, III функциональный класс ХСН по классификации New York Heart Association Functional Classification, фракция выброса ≤40%). Кровь из локтевой вены забирали в пробирки системы «Vacutainer» c Na-гепарином и затем получали фракцию нейтрофилов с использованием фиколла, как описано в [11]. Изолированные нейтрофилы ресуспендировали в полной культуральной среде DMEM, содержащей 10% термоинактивированной эмбриональной телячьей сыворотки, а также пенициллин (100 ЕД/мл), сульфат стрептомицина (100 мг/мл) и глутамин 2,0 мМ. Чистота нейтрофилОВ составила 98%, жизнеспособность – 99%.
Образование АФК нейтрофилами измеряли методом хемилюминесценции [11].
Для выявления путей внутриклеточной сигнализации, участвующих в механизмах действия исследуемых адреноблокаторов, клетки инкубировали с ингибиторами фосфатидил инозитол 3 киназы (PI3K) (LY294002 и Wortmannin), экстраклеточных регуляторных киназ (ERK) (PD98059), митогенактивируемой p38 киназы (p38MAPK) 30 минут. Затем к клеткам добавляли адреноблокаторы и в конце – LPS. Апоптотическую гибель нейтрофилов определяли, как описано ранее [12]. Статистическую обработку данных проводили в программе SigmaPlot.
Результаты исследования и их обсуждение
Первоначально было изучено воздействие пропранолола и фентоламина на уровень АФК, генерируемых нейтрофилами (рис. 1). Полученные результаты показали, что пропранолол в диапазоне концентраций от 25 до 400 мкМ дозозависимо увеличивал продукцию АФК (начиная с концентрации 50 мкМ). При концентрации пропранолола 200–400 мкМ продукция АФК нейтрофилами достигала максимальных значений (300%) (рис. 1а), в то время как активация продукции АФК нейтрофилами при действии LPS составляла 250% (рис. 1б). В присутствии LPS с ростом концентрации пропранолола происходило еще большее увеличение генерации АФК (~3,5 раза при концентрации от 100 до 400 мкМ) нейтрофилами, начиная с концентрации 50 мкМ пропранолола (рис. 1б). Однако отношение величины ХЛ в присутствии LPS к величине ХЛ без LPS с ростом концентрации пропранолола уменьшалось, достигая величины 1,17 (по сравнению с соотношением в отсутствие пропранолола, равным 2,5) (рис. 1а, 1б).
В отличие от пропранолола, фентоламин в диапазоне концентраций от 0,1 до 400 мкМ дозозависимо снижал продукцию АФК нейтрофилами, начиная с концентрации 20 мкМ, достигая минимальных значений при концентрации фентоламина 200–400 мкМ (рис. 1в). В присутствии LPS с ростом концентрации фентоламина происходило уменьшение генерации АФК нейтрофилами, начиная с концентрации 10 мкМ фентоламина (рис. 1г). Однако отношение величины ХЛ в присутствии LPS к величине ХЛ в отсутствие LPS с ростом концентрации фентоламина сохранялось (2,27 раза при концентрации 400 мкМ фентоламина) (рис. 1в, 1г).
Во второй серии экспериментов для выявления определяющих механизмов действия адреноблокаторов было изучено влияние специфических ингибиторов сигнальных путей активации клеток при действии липополисахаридов на генерацию АФК нейтрофилами в присутствии пропранолола и фентоламина. Было установлено, что SB203580 (в присутствии и отсутствие LPS) редуцировал уровень АФК нейтрофилов почти до уровня АФК контроля (рис. 2, SB). PD98059 ~ на 10% редуцировал уровень АФК нейтрофилов в присутствии пропранолола и LPS (рис. 2, PD). Ингибиторы фосфатидил инозитол 3 киназы (Wortmannin и LY294002) снижали уровень АФК контрольных и активированных LPS нейтрофилов (рис. 2, LY, W, а, б). При этом отношение величины уровней АФК в присутствии LPS к величине уровней АФК без LPS для Wortmannin и LY294002 было меньше 2. Причем у Wortmannin эта величина была меньше, чем у LY294002, и составляла ~ 1,5. В присутствии пропранолола также происходило значительное снижение продукции АФК (рис. 2, W, в, г) при действии этих ингибиторов.
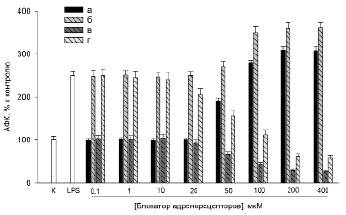
Рис. 1. Воздействие блокаторов адренорецепторов на уровень АФК, продуцируемых нейтрофилами при действии LPS. а, б – пропранолол; в, г – фентоламин; а, в – нейтрофилы без LPS; б, г – нейтрофилы вместе с LPS (20 нг/мл LPS). n=12, p<0,005
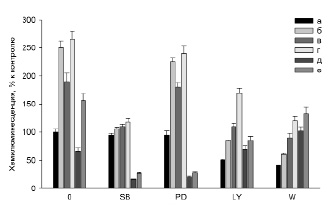
Рис. 2. Действие ингибиторов сигнальных путей на уровень АФК, генерируемых нейтрофилами, при добавлении к клеткам 50 мкМ пропранолола (Pro), 50 мкМ фентоламина (Phe) и LPS; а – пробы без Pro, Phe и LPS; б – пробы с 20 нг/мл LPS; в – пробы с Pro; г – пробы с Pro и 20 нг/мл LPS; д – пробы с Phe; е – пробы с Phe и 20 нг/мл LPS; 0 – пробы без ингибиторов. SB – 0,5 мкМ SB203580; PD – 10 мкМ PD98059; LY – 1 мкМ LY 294002; W – 10 нМ Wortmannin. n=12, p<0,005
Ингибиторы р38MAPK и ERK значительно редуцируют уровень АФК нейтрофилов в отсутствие LPS (~ в 3–4 раза) и в присутствии LPS (~ в 5–6 раз) (рис. 2, SB, PD, д, е). Ингибиторы PI3K не ингибируют действие фентоламина на контрольные клетки и незначительно снижают уровень АФК нейтрофилов в присутствии LPS (рис. 2, LY, W, д, е).
Таким образом, полученные результаты показали, что среди исследованных ингибиторов сигнальных путей наиболее значимо уровень АФК нейтрофилов в присутствии пропранолола снижают ингибиторы p38MAPK и PI3K. Ингибитор ERK не снижает уровень АФК нейтрофилов как в присутствии, так и в отсутствие LPS (рис. 2, PD, в, г сравнить с рис. 2, 0, в, г). В присутствии фентоламина наиболее значимо уровень АФК нейтрофилов снижают ингибиторы p38MAPK ERK. Ингибиторы PI3K действуют менее эффективно.
В третьей серии экспериментов было исследовано действие адреноблокаторов на апоптоз нейтрофилов. Полученные результаты показали, что пропранолол вызывает значительное увеличение процента апоптотических нейтрофилов по сравнению с контролем. Пропранолол уменьшает ингибирование апоптоза нейтрофилов, индуцированное LPS. Фентоламин незначительно увеличивает апоптоз нейтрофилов (рис. 3).
Ответ нейтрофилов на эндотоксины можно условно разделить на три стадии. На начальной стадии вслед за активацией TLR4 – сигнального пути происходит активация НАДФН-оксидазы, затем в течение нескольких часов развивается секреция цитокинов, в более поздней стадии проявляются изменения в регуляции апоптоза клеток. Наши результаты показали, что фентоламин значительно снижает уровень АФК, генерируемых нейтрофилами в присутствии LPS (рис. 1в, 1г). Первым из цитокинов обычно секретируется TNFα. В экспериментах на мышиных макрофагах пропранолол и фентоламин не снижали LPS-индуцированную продукцию TNFα, но несколько снижали продукцию этого цитокина при совместном действии норадреналина и LPS. Показано, что ингибиторы JNK, ERK, p38MAPK могут частично снижать секрецию TNFα, индуцированную норадреналином [13].
Известно, что в активации секреции TNFα моноцитами человека при действии LPS ключевую роль играет путь PI3K→Akt (RAC-alpha serine/threonine-protein kinase, Protein kinase B alpha), который может инактивировать сигнальные пути с участием p38MAPK, JNK и ERK [5]. В наших экспериментах основное различие в сигнальных путях пропранолола и фентоламина приходится на ERK. Для фентоламина ингибиторы LY и Wortmannin значительно слабее ингибируют пробы (рис. 3, LY294002, д, е; рис. 3, W, д, е) по сравнению с аналогичными пробами при действии SB (рис. 3, SB, д, е). Это может говорить о некоторой отрицательной регуляции для фентоламина по этим сигнальным путям.
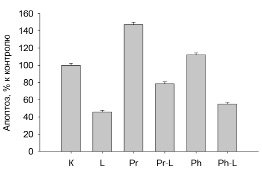
Рис. 3. Влияние пропранолола, фентоламина и LPS на апоптоз нейтрофилов. К – контроль, L – 100 нг/мл LPS, Pr – 200 мкМ пропранолола, Pr-L – последовательное добавление к клеткам пропранолола и LPS, Ph – 200 мкМ фентоламин, Ph-L последовательное добавление к клеткам фентоламина и LPS. n=12, p<0,005
Кроме того, LY294002 ингибирует только PI3K, а Wortmannin – PI3K и Akt. В экспериментах на фибробластах животных было показано, что фентоламин эффективно снижает секрецию цитокинов: IL-1β, IL-6 и IL-8 [14], индуцированную LPS.
Одним из механизмов активации апоптоза нейтрофилов являются АФК. Наши эксперименты показали, что пропранолол увеличивает продукцию АФК нейтрофилами, что, возможно, является одной из причин увеличения апоптоза этих клеток в наших экспериментах. Также пропранолол, увеличивая апоптотическую гибель нейтрофилов в присутствии LPS, будет способствовать снижению времени циркуляции этих клеток при воспалительных патологиях. Фентоламин оказывает на апоптоз менее выраженное действие, чем пропранолол, однако он эффективно снижает продукцию АФК этими клетками в присутствии LPS. Вероятно, фентоламин активирует апоптоз путем снижения мембранного потенциала митохондрий с последующей активацией каспаз, как это было показано в некоторых типах клеток [15].
Заключение
Полученные результаты показали, что пропранолол увеличивает уровень АФК в диапазоне концентраций от 50 мкМ до 400 мкМ. В присутствии ингибиторов p38MAPK и PI3K происходит значительное снижение пропранолол-индуцированной продукции АФК нейтрофилами, что указывает на участие этих ферментов в активации клеток под действием пропранолола. В отличие от пропранолола, фентоламин значительно снижает продукцию АФК клетками в диапазоне концентраций от 20 мкМ до 400 мкМ. В механизме действия пропранолола большое значение имеют p38MAPK и PI3K. В механизме действия фентоламина значимая роль принадлежит p38MAPK, ERK, при этом действие PI3K менее выражено.
Работа выполнялась в рамках госзадания по теме 0576-2020-0005, одобрена ЛЭК БПНЦ РАН, протокол № 14 от 04.09.2018 г.
Библиографическая ссылка
Юринская М.М., Винокуров М.Г., Сусликов А.В. АДРЕНОБЛОКАТОРЫ ПРОПРАНОЛОЛ И ФЕНТОЛАМИН МОДУЛИРУЮТ LPS-ИНДУЦИРОВАННУЮ ВНУТРИКЛЕТОЧНУЮ СИГНАЛИЗАЦИЮ НЕЙТРОФИЛОВ ЧЕЛОВЕКА // Международный журнал прикладных и фундаментальных исследований. 2022. № 11. С. 7-12;URL: https://applied-research.ru/en/article/view?id=13460 (дата обращения: 01.07.2026).
DOI: https://doi.org/10.17513/mjpfi.13460