
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
RECOMBINANT EXPRESSION OF T7 RNA POLYMERASE IN E. COLI STRAIN FOR RNA SYNTHESIS IN VITRO

Синтез одноцепочечных молекул РНК in vitro – широко используемая лабораторная процедура, которая активно используется как для научных задач по исследованию РНК, так и для получения терапевтических препаратов на основе РНК. Этот метод позволяет вносить модификации в РНК, а также адаптировать синтез РНК под разные задачи. Так, этот способ применим для биохимического и молекулярного анализа РНК, а также структурного анализа комплексов между РНК и белками. Кроме того, использование синтезированной in vitro РНК сыграло важную роль в разработке мРНК-вакцин, инструментов редактирования генома CRISPR/Cas9 и создании плюрипотентных стволовых клеток [1–5].
Основными ферментами для синтеза РНК in vitro являются ДНК-зависимые РНК-полимеразы вирусов бактерий (Т7, Т3, SP6), среди которых фермент из Т7 бактериофага (Т7РНКП) является самым распространенным в практическом применении. Эти ферменты позволяют синтезировать РНК как в лабораторных условиях, так и на производстве. В качестве матриц для синтеза могут быть использованы либо фрагменты ДНК, полученные в ходе ПЦР, либо линеаризованные плазмиды, содержащие целевую последовательность под контролем Т7-промотора. Использование Т7РНКП для синтеза РНК позволяет кепировать ее искусственными кеп-аналогами в процессе ко-транскрипционного кепирования [6]. Однако для масштабирования синтеза РНК in vitro и воспроизведения технологий, лежащих в основе производства мРНК-вакцин, необходимо получить штамм-продуцент Т7РНКП для получения работоспособных препаратов этого фермента. Ранее были предложены схемы продукции Т7РНКП на основе коммерческих векторов, где индукция осуществляется с помощью ИПТГ, что экономически невыгодно при переходе к большим объемам на ферментерах [7–9]. Поэтому для масштабного получения этого фермента необходим бактериальный штамм-продуцент с подходящим селекционным маркером и недорогим индуктором. В качестве экспериментальной попытки улучшить процессивность Т7РНКП мы модифицировали фермент, получив химерный белок, состоящий из Т7РНКП и белка Sso7d из Sulfolobus solfataricus.
В данной работе мы получили штамм-продуцент E. coli, несущий экспрессионный вектор для высокой продукции Т7РНКП, а также подобрали оптимальные условия экспрессии и очистки этого фермента для дальнейшего использования в отработке технологии производства терапевтических препаратов на основе мРНК. Также мы создали химерный белок, состоящий из белка Sso7d, повышающего процессивность ДНК-связывающих белков, и Т7РНКП, чтобы далее исследовать влияние на продуктивность синтеза РНК in vitro.
Материалы и методы исследования
Получение генетических конструкций
Нуклеотидные последовательности, кодирующие Т7РНКП и белок Sso7d, синтезировали de novo из олигонуклеотидов. Далее фрагменты ДНК, соответствующие Т7РНКП и белку Sso7d, амплифицировали ПЦР с использованием высокоточной ДНК-полимеразы Q5 (NEB) и олигонуклеотидов, содержащих уникальные сайты рестрикции NdeI и NotI (NEB). Также во время наработки ДНК-фрагмента ПЦР добавляли нуклеотидную последовательность, кодирующую шесть гистидинов (6His) на С-конце Т7РНКП для дальнейшей аффинной очистки фермента. После этого фрагменты были очищены через агарозный гель набором для экстракции из агарозы (Евроген). Очищенные препараты ДНК-фрагментов были использованы в реакции рестрикции обеими эндонуклеазами рестрикции (37 °С, 1 ч), после чего следовала стадия переосаждения ДНК (3М ацетат натрия, 96 % этанол) центрифугированием на максимальных оборотах настольной миницентрифуги (Eppendorf Minispin). Экспрессионный вектор pSOL (Lucigen) также гидролизовали двумя эндонуклеазами рестрикции NdeI и NotI (NEB) в течение 3 ч, а далее очищали через агарозу. Реакцию лигирования проводили при 22 °С в течение 1 ч при помощи ДНК-лигазы Т4, а затем лигазной смесью трансформировали компетентные клетки E. coli 10G (Lucigen). Чашки Петри с агаризованной средой LB и канамицином инкубировали в течение 12 ч при 37 °С до появления единичных колоний. На следующий день проводили ПЦР анализ отдельных колоний на содержание вставки, из положительных колоний выделяли плазмидную ДНК и секвенировали. Плазмиды с корректными вставками нарабатывали, выделяли и трансформировали экспрессионные штаммы E. coli, предназначенные для продукции рекомбинантных белков.
Экспрессия Т7РНКП в клетках E. coli
Для проведения экспериментов по экспрессии рекомбинантной Т7РНКП использовали две микробиологические среды: LB (10 г/л триптон, 5 г/л дрожжевой экстракт, 5 г/л хлорид натрия) и 2YТ (16 г/л триптон, 10 г/л дрожжевой экстракт, 5 г/л хлорид натрия). Для проверки экспрессии использовались следующие штаммы: KRX (Promega); Rosetta gami 2 (Novagen); BL21 DE3 (Invitrogen); E. cloni 10G (Lucigen). Ниже приведены генотипы этих штаммов.
E. cloni 10G: F- mcrA Δ(mrr-hsdRMS-mcrBC) endA1 recA1 Φ80dlacZΔM15 ΔlacX74 araD139 Δ(ara,leu)7697galU galK rpsL nupG λ- tonA (StrR)
BL21(DE3): F- ompT hsdSB (rB-mB-) gal dcm (DE3)
Rosetta gami 2: Δ(ara-leu)7697 ΔlacX74 ΔphoA PvuII phoR araD139 ahpC galE galK rpsL (DE3) F′[lac+ lacIq pro] gor522::Tn10 trxB pRARE2 (CamR, StrR, TetR)
KRX: [F´, traD36, ΔompP, proA+B+, lacIq, Δ(lacZ)M15] ΔompT, endA1, recA1, gyrA96 (Nalr), thi-1, hsdR17 (rk–, mk+), e14– (McrA–), relA1, supE44, Δ(lac-proAB), Δ(rhaBAD)::T7 RNA polymerase.
Реципиентные штаммы E. coli, предназначенные для продукции рекомбинантных белков трансформировали полученными генетическими конструкциями. Трансформационную смесь высевали на чашки Петри с агаризованной средой и селективным антибиотиком и инкубировали ночь при 37 °С до появления единичных колоний. Далее единичной колонией инокулировали 5 мл жидкой питательной среды с антибиотиком (канамицин, 30 мкг/мкл) и растили ночь при 37 °С на шейкере (Eppendorf Innova, 180 об/мин). На следующий день ночной культурой заражали жидкую питательную среду в качалочных колбах (1/10 объема) и подращивали при 37 °С до оптической плотности OD600 = 0,4–0,6 единиц. Далее в колбы добавляли рамнозу (0,05–0,2 %) в качестве индуктора экспрессии и инкубировали клеточную культуру при разных температурах (от 28 °С до 37 °С) до оптической плотности OD600 = 1–1,2. Клетки осаждали центрифугированием при 5000 g в течение 30 мин при 4 °С.
Выделение и очистка Т7РНКП
Биомассу ресуспендировали в буфере для нанесения 50 mM калий-фосфатного буфера рН 7,6, 300 мМ NaCl, 5mM имидазола на льду. Далее бактериальные клетки разрушали ультразвуком во льду до просветления суспензии. Полученный лизат центрифугировали (10000 g, 40 мин при 4 °С) до осаждения клеточного дебриса и визуального просветления супернатанта. Супернатант осторожно отбирали и хранили на льду до нанесения на хроматографическую колонку. Аликвоту супернатанта и клеточного дебриса отбирали для дальнейшего электрофоретического анализа. Полученный супернатант наносили на колонку с сорбентом Ni-NTA, предварительно промытую буфером (50 mM калий-фосфатного буфера рН 7,6, 300 mМ NaCl, 5 mM имидазола). Связавшийся с сорбентом белок элюировали градиентом концентрации имидазола (25–500 mМ) в том же буфере. Фракции, содержащие максимальный уровень белка, объединяли и диализовали против буфера для хранения (50 mM Tris-HCl pH 8,0, 100 mM NaCl) с глицерином на -20 °С. Для избежания деградации РНК рибонуклеазами все растворы готовились на воде, обработанной DEPC. Электрофорез в полиакриламидном геле (ПААГ) проводили по стандартной методике Лэммли. Для белкового электрофореза в денатурирующих условиях использовался 8–12 % ПААГ. К белковому препарату добавлялся двукратный объем буфера для нанесения (250 mМ Трис-HCl pH 6,8, 6 % SDS, 2 % меркаптоэтанол, 16 % глицерин, 0,05 % бромфеноловый синий), раствор тщательно перемешивали и выдерживали в кипящей водяной бане в течение 5 мин. Окраску геля проводили с использованием Кумасси бриллиантового синего R-250. Для электрофореза использовали в трис-глициновый буфер (10х 1 % SDS 0,25 М Трис-OH и 1,8 М глицин) на приборе фирмы Bio-Rad (CША).
Постановка реакции транскрипции in vitro
ДНК-матрицу, содержащую последовательность Т7-промотора (ДНК-фрагмент после ПЦР или линеаризованная плазмида), перед постановкой реакции очищали через агарозный гель набором для экстракции из агарозы (Евроген). Реакционная смесь объемом 20 мкл содержала следующие компоненты: 50 нг/мкл ДНК, 2 mM rNTPs, 1x транскрипционный буфер (40 mM TrisHCl pH 8,6 mM MgCl2, 5 mM DTT), 0,2 мкг/мкл Т7РНКП. Реакцию транскрипции in vitro проводили при 37 °С в течение 1 ч. Продукты ферментативной реакции анализировали при помощи агарозного электрофореза в денатурирующих условиях.
Результаты исследования и их обсуждение
Использование искусственных мРНК в терапевтических целях имеет огромный потенциал, что показало недавнее широкое применение мРНК вакцин [1, 2]. Синтез РНК in vitro в лабораторных и промышленных условиях осуществляется ДНК-зависимыми РНК-полимеразами бактериофагов, в частности бактериофага Т7. Фермент Т7РНКП является односубъединичным глобулярным белком с молекулярной массой около 100 кДа. Этот фермент специфично связывается с последовательностью ДНК Т7-промотора (23 нуклеотида), что обеспечивает дальнейший синтез РНК с ДНК-матрицы in vitro. Таким способом могут быть синтезированы молекулы РНК длиной от нескольких десятков до нескольких тысяч нуклеотидов. Продуктивность таких реакций транскрипции in vitro может достигать миллиграммовых количеств. Оптимизация реакции транскрипции in vitro является важной задачей как с точки зрения повышения качества итоговой РНК, так и продуктивности самой реакции [10]. Ранее было показано, что белок Sso7d из Sulfolobus solfataricus, ковалентно сшитый с ДНК-полимеразами, увеличивает их процессивность in vitro [11]. Авторы утверждают, что подобная стратегия по увеличению сродства к ДНК может быть применима к любому ДНК-связывающему ферменту, поэтому мы решили получить химерный белок, в котором Sso7d и Т7РНКП связаны гибким линкером, для дальнейшего исследования влияния Sso7d на процессивность Т7РНКП и продуктивность синтеза РНК in vitro.
Для создания штамма-продуцента T7РНКП и химерного белка Sso7d-T7РНКП была использована коммерческая экспрессионная система, контроль над продукцией рекомбинантных белков в которой происходит за счет рамнозного промотора (pSOL, Lucigen). Благодаря этому промотору можно проводить тонкую настройку экспрессии рекомбинантного белка с помощью титрования рамнозой. Также использование этой экспрессионной системы позволяет снизить количество белковых молекул с неправильной укладкой, которые в итоге формируют нерастворимые агрегированные частицы (тельца включения). Это свойство экспрессионного вектора pSOL дает преимущество при продукции сложных белковых молекул, склонных к агрегации, например, таких как химерные белки из разных полипептидных цепей, соединенных ковалентно. Одновременно с этим вектор pSOL имеет малый размер и подходящий для крупных наработок фермента селекционный маркер (канамицин). Дополнительные терминаторы транскрипции предотвращают сквозную транскрипцию в плазмиде, что дополнительно стабилизирует вставку.
На первом этапе работы мы синтезировали ДНК-последовательности, кодирующие Т7РНКП и белок Sso7d de novo из олигонуклеотидов. Далее при помощи ПЦР мы синтезировали последовательность ДНК, кодирующую химерный белок Sso7d-Т7РНКП, где две полипептидные цепи были соединены через гибкий аминокислотный мостик (GGGSGGGSGGG). Итоговый расчетный размер химерного белка составляет 108 кДа. После этого полученные фрагменты ДНК были клонированы в экспрессионный вектор pSOL по уникальным сайтам, как описано выше. В результате были получены генетические конструкции, кодирующие Т7РНКП (рис. 1, а) и химерный белок Sso7d-Т7РНКП (рис. 1, б) под контролем рамнозного промотора.
Далее мы провели сравнительный анализ продукции обоих белков в четырех разных экспрессионных штаммах E. coli, оптимизированных для получения рекомбинантных белков: KRX (Promega); Rosetta gami 2 (Novagen); BL21 DE3 (Invitrogen); E. cloni 10G (Lucigen). По результатам пробных наработок ферментов лучший результат был в штамме BL21 DE3 (рис. 2).
Остальные штаммы показали значительно более низкую продукцию белков, поэтому дальнейшая отработка условий наработки ферментов проводилась на штамме BL21 DE3. Итоговые условия для лучшей продукции обоих функционально-активных ферментов включают такие факторы, как: а) среда для культивирования 2YТ; б) температура культивирования до индукции 37 °С, температура культивирования после индукции 37 °С; в) оптическая плотность культуры до индукции 0,5–0,6 ОЕ, а концентрация рамнозы в качестве индуктора – 0,2 %; г) экспрессионный штамм BL21 DE3. Типовой результат оптимальной продукции этих белков показан на рис. 3.
Очистка Т7РНКП осуществлялась с помощью аффинного сорбента Ni-NTA (Qiagen). Бактериальные клетки после осаждения из культуральной жидкости ресуспендировали в буфере для нанесения на хроматографическую колонку и разрушали с помощью ультразвука, а полученную суспензию осветляли центрифугированием. После этого супернатант наносили на хроматографическую колонку с сорбентом и элюировали связавшийся белок градиентом концентрации имидазола. Все процедуры детально описаны в разделе «Материалы и методы исследования». Точно так же выделяли химерный белок Sso7d-T7РНКП. В условиях эксперимента химерный белок практически весь находился в растворимой фракции, что может указывать на корректный фолдинг как белка Sso7d, так и Т7РНКП в составе одной полипептидной цепи. Таким образом, можно предполагать, что выбранный гибкий линкер позволяет двум белкам соседствовать в пространстве. После электрофоретического анализа собранных фракций часть из них объединялась и переводилась в буфер для хранения диализом. Полученные препараты имели чистоту от 90 до 95 % и итоговый выход продуктов от 35 мг/л до 40 мг/л (рис. 3, а и б).
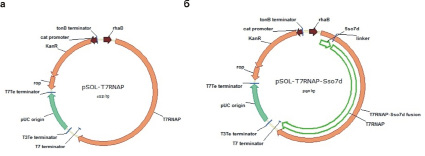
Рис. 1. а) Карта экспрессионного вектора для продукции Т7РНКП под контролем рамнозного промотора; б) Карта экспрессионного вектора для продукции химерного белка Sso7d-Т7РНКП под контролем рамнозного промотора
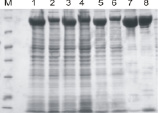
Рис. 2. Сравнительный анализ наработки белков Т7РНКП (100 кДа) и Sso7d-Т7РНКП (108 кДа) в четырех штаммах E. coli. Результат электрофореза в ПААГ в денатурирующих условиях, 5 мкл бактериальной культуры + 10 мкл буфера для нанесения (1–2 – Т7РНКП и Sso7d-Т7РНКП в штамме KRX; 3–4 – Т7РНКП и Sso7d-Т7РНКП в штамме Rosetta gami 2; 5–6 – Т7РНКП и Sso7d-Т7РНКП в штамме E. cloni 10G; 7–8 – Т7РНКП и Sso7d-Т7РНКП в штамме BL21 DE3
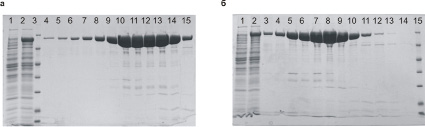
Рис. 3. а) Наработка и очистка Т7РНКП (1 – проскок после нанесения на колонку, 2 – наносимый образец, 3 – маркер, 4–15 элюция градиентом имидазола). б) Наработка и очистка химерного белка Sso7d-Т7РНКП (1 – проскок после нанесения на колонку, 2 – наносимый образец, 3–14 элюция градиентом имидазола, 15 – маркер)
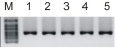
Рис. 4. Электрофоретический анализ в агарозном геле продуктов реакции транскрипции in vitro с использованием коммерческого фермента Т7РНКП Thermo Scientific (1), выделенного препарата Т7РНКП (2–0,1 мкг/мкл фермента; 3–0,2 мкг/мкл фермента) и препарата химерного белка Sso7d-Т7РНКП (4–0,1 мкг/мкл фермента; 5–0,2 мкг/мкл фермента)
Следующим этапом была проведена проверка активности полученных ферментов. Для этого при помощи ПЦР была получена ДНК-матрица, содержащая Т7-промотор. После очистки фрагментов ДНК с использованием электрофореза в агарозном геле их использовали в реакции транскрипции in vitro, как описано в разделе «Материалы и методы исследования». Выделенная Т7РНКП показала схожую активность в сравнении с коммерческим препаратом Т7РНКП от компании Thermo Scientific. Химерный белок Sso7d-T7РНКП также продемонстрировал активный синтез РНК in vitro, однако каких-то существенных преимуществ относительно Т7РНКП не дал, по крайней мере в условиях предварительного эксперимента (рис. 4).
Ранее было показано, что для химер Sso7d и ДНК-полимераз наибольший преимущественный эффект наблюдался при синтезе ампликонов длиной до 10000 нуклеотидов [11–14], в нашем же случае использовались относительно короткие ДНК-матрицы (от 500 до 2000 нуклеотидов), что могло нивелировать разницу между процессивностью Т7РНКП дикого типа и гибридного белка Sso7d-Т7РНКП. Также возможно, что для демонстрации потенциального преимущества химерного белка необходима оптимизация самой реакции синтеза РНК (концентрация ионов магния, концентрация нуклеотидов, время инкубации), что может быть предметом следующего исследования.
Заключение
В данной работе был получен бактериальный продуцент Т7РНКП на основе штамма E. coli и экспрессионного вектора, подходящих для масштабных наработок этого фермента. В ходе экспериментов подобрали оптимальные условия для продукции и очистки Т7РНКП. Полученный препарат фермента по активности не уступает коммерческому и может быть использован в реакции синтеза РНК in vitro как для исследовательских задач в лабораторной практике, так и для терапевтического применения при изготовлении мРНК-вакцин. Также мы модифицировали фермент Т7РНКП в целях повышения синтеза РНК in vitro при помощи создания химерного белка с Sso7d из Sulfolobus solfataricus, однако подбор оптимальных условий реакции для этого гибридного белка требует дальнейших экспериментов.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Работа выполнена при поддержке программы Министерства высшего образования и науки РФ (соглашение № 075-10-2021-113, уникальный номер проекта RF-193021X0001).
Библиографическая ссылка
Захарова М.В., Нагорных М.О. ПОЛУЧЕНИЕ БАКТЕРИАЛЬНОГО ПРОДУЦЕНТА ДНК-ЗАВИСИМОЙ РНК-ПОЛИМЕРАЗЫ БАКТЕРИОФАГА Т7 ДЛЯ СИНТЕЗА РНК IN VITRO // Международный журнал прикладных и фундаментальных исследований. 2022. № 12. С. 9-14;URL: https://applied-research.ru/en/article/view?id=13476 (дата обращения: 27.06.2026).
DOI: https://doi.org/10.17513/mjpfi.13476