
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
VALIDATION OF METHODS FOR DETERMINING THE COMPOSITION OF FATTY ACIDS IN PHARMACY

Введение
Жирные кислоты и их производные находят широкое применение в фармации. Некоторые из них обладают своей собственной фармакологической активностью [1-3]. Например, полиненасыщенные жирные кислоты и некоторые виды растительных масел. Среди вспомогательных веществ в фармацевтической технологии имеется внушительное количество жирных кислот и их производных, которые обладают эмульгирующими, скользящими, противоприлипающими и пластифицирующими свойствами.
Для подтверждения подлинности и количественного определения примесей при исследовании подобного рода веществ применяется метод газовой хроматографии [4; 5]. Согласно рекомендациям XV издания Государственной фармакопеи (ГФ) и международных стандартов, любую аналитическую методику необходимо подвергать валидации, что выражается в экспериментальном подтверждении пригодности таковой к практическому использованию в лаборатории в плане обеспечения получения достоверной информации об объекте исследования [6; 7].
Но методики исследования состава жирных кислот методом газовой хроматографии используются в практике лабораторий с середины прошлого века. С тех пор происходило постоянное усовершенствование газовых хроматографов и программного обеспечения, а также появление новых подходов к обработке хроматограмм и современных требований к достоверности результатов испытаний. А потому проведение валидации методики определения состава жирных кислот с использованием современного оборудования и программного обеспечения представляется актуальной задачей.
Целью исследования является обоснование выбора метода дериватизации стеарата магния для дальнейшего исследования методом газовой хроматографии и оптимизация условий хроматографического разделения. Посредством исследования намечено доказать повторяемость, внутрилабораторную прецизионность, робастность и правильность. Задачей ставится оценка предела обнаружения, предела количественного определения и аналитической области методики.
Материалы и методы исследования
Валидация методики проводится с использованием газового хроматографа Shimadzu 2010 в комплекте с пламенно-ионизационным детектором. Образцы европейской фармакопеи пальмитиновой и стеариновой кислот выступают в качестве стандартных. Испытуемым образцом является произведенный в Нидерландах стеарат магния. А для хроматографического разделения используется капиллярная колонка DB-WAX, Agilent, которая изготовлена из плавленого диоксида кремния, имеет длину 30 м и диаметр 0,32 мм, а также нанесенную фазу макрогол 20000 P с толщиной пленки в 0,5 мкм.
Результаты исследований и их обсуждение
Дериватизация используется в анализе состава жирных кислот с целью получения более летучих дериватов. Жирные кислоты за счет высокой молекулярной массы и наличия гидроксильных полярных групп, склонных к образованию водородных связей, обладают низкой летучестью. Алкилирование, по результатам которого получают метиловые, этиловые и пропиловые эфиры карбоновых кислот, чаще всего применяют для исследования жирных кислот и их производных методом газовой хроматографии.
Сложные эфиры из свободных жирных кислот получают этерификацией, а в случае с триацилглицеридами и прочими производными жирными кислотами используется переэтерификация [8; 9]. Однако в щелочных условиях свободные жирные кислоты не этерифицируются, поэтому переэтерификация не подходит для триацилглицеридов с их содержанием. А переэтерификация и этерификация в последовательных щелочных и кислотных условиях пригодна для всех триглицеридов, которые не содержат лауриновые жирные кислоты [10]. Для проб с жирными кислотами целесообразно применять метод этерификации в кислотных условиях. Метанольный раствор трифторида бора этерифицирует свободные жирные кислоты и переэтерифицирует сложные эфиры жирных кислот [11]. И поэтому для получения метиловых эфиров из стеарата магния и используемых в качестве стандартных образцов пальмитиновой и стеариновой кислот выбран трифторид бора в метаноле (14%-ный раствор).
Объектом валидации выступает методика определения состава жирных кислот во вспомогательном веществе «магния стеарат» в соответствии с монографией 01/2022-0229 из 11 издания Европейской фармакопеи [12]. По итогам работы внесены некоторые уточнения в методику. Так, установлено, что деление потока газа-носителя 1 к 20 способствует отсутствию перегрузки капиллярной колонки и соблюдению требований к пригодности хроматографической системы. Перед валидацией важно проводить проверку пригодности хроматографической системы на соответствие требованиям методики, результаты которой представлены в таблице 1. Высокое значение разрешения между пиками метилпальмитата и метилстеарата указывает на то, что методика специфична.
Примеры хроматограмм представлены на рисунках 1 и 2.
В процессе валидации доказана линейность методики в широком диапазоне концентраций растворов стандартных образцов по 10 точкам от предела обнаружения методики до 120% от максимальной концентрации жирной кислоты в пробе. Рассчитанные коэффициенты корреляций составляют 0,9991 для пальмитиновой кислоты и 0,9993 для стеариновой кислоты, что выше требований ГФ РФ XV к коэффициенту 0,99. Построенные графики линейных зависимостей представлены на рисунках 3 и 4. Свободные члены линейных зависимостей ничтожно малы. Ниже допустимого уровня шума прибора 100 мкВ.
В процессе валидации удалось установить внутрилабораторную прецизионность методики посредством оценки значимости различий дисперсий двух выборок, полученных двумя разными операторами после проверок данных выборок на однородность [13-15]. Каждая выборка состояла из 10 повторностей. Расчетный критерий Фишера в данном случае ниже табличного значения. Также оценена робастность методики при замене газа-носителя с гелия на азот. Результаты приведены в таблице 2.
Таблица 1
Проверка пригодности хроматографической системы
|
Критерии приемлемости |
Требования |
Результат |
|
Время удерживания пика метилпальмитата относительно пика метилстеарата |
около 0,9 |
0,8846 |
|
Разрешение между пиком метилпальмитата и пиком метилстеарата на хроматограмме стандартного раствора |
не менее 5,0 |
43,0 |
|
Относительное стандартное отклонение для площадей пиков метилпальмитата по результатам 6 повторных инжекций стандартного раствора |
не более 3,0% |
1,8% |
|
Относительное стандартное отклонение для площадей пиков метилстеарата по результатам 6 повторных инжекций стандартного раствора |
не более 3,0% |
1,4% |
|
Относительное стандартное отклонение для отношения площадей пиков метилпальмитата и метилстеарата по результатам 6 повторных инжекций стандартного раствора |
не более 1,0% |
0,4% |
Источник: составлено авторами по результатам данного исследования.
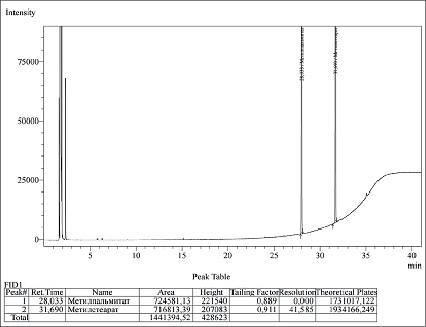
Рис. 1. Единичная хроматограмма стандартного раствора Источник: составлено авторами по результатам данного исследования
Выявленные в ходе валидации характеристики методики приведены в таблице 3. Для определения предела обнаружения и предела количественного определения использовался метод инструментальной оценки. В процессе валидации установлена концентрация раствора стандартных образцов, которая образует пик на хроматограмме с отношением уровня сигнала к шуму, равным 3. Данная концентрация (0,0125% от суммы жирных кислот) является пределом обнаружения. Концентрация раствора стандартных образцов, которая образует пик на хроматограмме с отношением сигнала к шуму, равным 10, является пределом количественного определения (0,04% от суммы жирных кислот).
Таблица 2
Оценка значимости различий дисперсий двух выборок при оценке показателей «внутрилабораторная прецизионность», «робастность»
|
Наименование показателя |
Внутрилабораторная прецизионность |
Робастность |
||
|
Метилпальмитат |
Метилстеарат |
Метилпальмитат |
Метилстеарат |
|
|
Критерий Фишера табличный, F (0,05;9;9) |
3,18 |
|||
|
Критерий Фишера расчетный |
1,69 |
1,69 |
2,86 |
2,86 |
Источник: составлено авторами по результатам данного исследования.
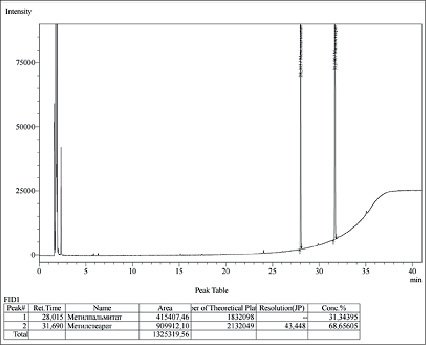
Рис. 2. Единичная хроматограмма испытуемого раствора Источник: составлено авторами по результатам данного исследования
Таблица 3
Показатели, установленные в результате валидации методики
|
Показатель повторяемости |
0,027% |
|
Показатель внутрилабораторной прецизионности |
0,076% |
|
Аналитическая область методики |
От 0,0125% до 100% от суммы жирных кислот |
|
Предел количественного определения |
0,04% жирной кислоты от суммы жирных кислот |
|
Предел обнаружения |
0,0125% жирной кислоты от суммы жирных кислот |
|
Неопределенность результатов исследования |
0,06% |
Источник: составлено авторами по результатам данного исследования.
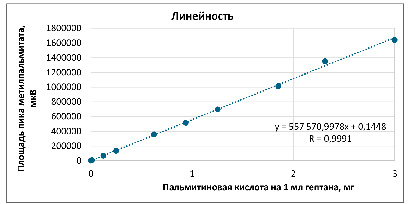
Рис. 3. График зависимости площадей пиков метилпальмитата от массы навески пальмитиновой кислоты Источник: составлено авторами по результатам данного исследования
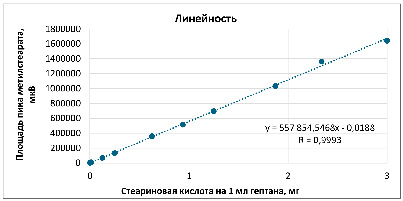
Рис. 4. График зависимости площадей пиков метилстеарата от массы навески стеариновой кислоты Источник: составлено авторами по результатам данного исследования
Заключение
В процессе исследования теоретически обосновано использование 14% раствора трифторида бора в метаноле для дериватизации стеарата магния, подобраны условия проведения хроматографического разделения и установлено значение деления потока газа-носителя 1:20. Проведена валидация методики исследования состава жирных кислот с использованием современных поверенных средств измерения. По итогам валидации определено, что методика характеризуется соблюдением линейности в широком диапазоне от 0,0125% до 120% и специфична, доказана ее правильность. Полученные результаты оценки повторяемости, внутрилабораторной прецизионности и робастности методики удовлетворяют критериям приемлемости.
Валидация методики исследования состава жирных кислот подтверждает собственную пригодность для получения достоверных сведений об испытуемом образце с высокой точностью. Установленный предел обнаружения 0,0125% от суммы жирных кислот оказался лучше значения в 1%, которое присуще для более ранних сведений о характеристиках подобных методик. Предел количественного определения составил 0,04%, что соответствует указаниям современных методик исследования состава жирных кислот, согласно которым не учитываются пики менее 0,05% от суммарного содержания жирных кислот.
Conflict of interest
Библиографическая ссылка
Трубицына И.М., Василенко И.А. ВАЛИДАЦИЯ МЕТОДИК ОПРЕДЕЛЕНИЯ СОСТАВА ЖИРНЫХ КИСЛОТ В ФАРМАЦИИ // Международный журнал прикладных и фундаментальных исследований. 2025. № 6. С. 33-38;URL: https://applied-research.ru/en/article/view?id=13734 (дата обращения: 03.08.2026).
DOI: https://doi.org/10.17513/mjpfi.13734