
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
SYNTHESIS AND IN SILICO STUDY OF THE PHARMACOLOGICAL ACTIVITY OF NEW N-(2-BENZYLOXY-2-OXOETHYL) DERIVATIVES OF NITROGEN-CONTAINING HETEROCYCLIC COMPOUNDS

Введение
Сложноэфирные производные азотсодержащих гетероциклических соединений представляют собой ценные синтоны для получения разнообразных фармакологически активных веществ. Высокая реакционная способность сложноэфирной группы по отношению к нуклеофильным агентам позволяет легко получать на их основе соединения амидного типа. В настоящее время на основе сложноэфирных производных пиримидиновых соединений осуществляется направленный синтез веществ, содержащих линейные и циклические гуанидиновые фрагменты в своей структуре. Соединения указанного типа проявили себя в качестве высокоэффективных ингибиторов натрий-водородного обменника первого типа (NHE-1) [1; 2]. Подобные соединения, содержащие фрагменты гуанидина в своей структуре, рассматриваются как потенциальные нейро- и кардиопротекторные лекарственные средства [3; 4].
Однако самостоятельная фармакологическая активность указанных промежуточных сложноэфирных производных азотсодержащих гетероциклических соединений исследована в недостаточной степени, и имеются лишь отдельные публикации на эту тему. В частности, описан 1-(2-бензилокси-2-оксоэтил)хиназолин-4(3Н)-он (1) (рис. 1), содержащий фрагмент бензилацетата в качестве заместителя в положении N3 и проявивший высокую активность в отношении золотистого стафилококка и ряда других возбудителей бактериальных инфекций in vitro [5; 6] при низкой острой токсичности in vivo [7].
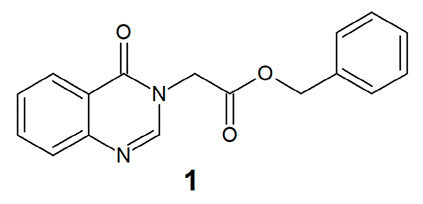
Рис. 1. 1-(2-Бензилокси-2-оксоэтил)хиназолин-4(3Н)-он Примечание: составлено авторами на основе источников [5; 6]
В связи с этим представляет существенный интерес синтез и прогностическое исследование in silico фармакологической активности других гетероциклических производных с аналогичным характером замещения при атомах азота.
Цели исследования – проведение направленного синтеза новых N-замещенных сложноэфирных производных азотсодержащих гетероциклических соединений и последующая прогностическая оценка их фармакологического потенциала с использованием методов in silico для выявления возможной биологической активности, в том числе противоопухолевой.
Материалы и методы исследования
Синтез целевых соединений осуществляли двумя методами:
1. Классическое N-алкилирование гетероциклов (бензимидазол, 2-оксибензимидазол, бензотриазол, аденин) бензилбромацетатом в среде диметилформамида в присутствии калия карбоната при комнатной температуре.
2. Метод «сплавления» триметилсилилпроизводных урацила, тимина и хиназолин-2,4(1H,3H)-диона с бензилбромацетатом при нагревании без растворителя (140–180°C) с удалением летучих продуктов в вакууме.
Структура синтезированных соединений подтверждена методами ЯМР-спектроскопии (¹H и ¹³C) на спектрометре Bruker Avance 600 в ДМСО-d₆.
Температуры плавления определены в капиллярах на приборе Mel-Temp 3.0.
Контроль чистоты проводили методом тонкослойной хроматографии на пластинах Silica gel 60 F₂₅₄.
Прогноз выполнен с использованием веб-сервиса PASS Online, который оценивает вероятности биологических эффектов на основе анализа молекулярной структуры.
Результаты исследований и их обсуждение
Наиболее простым и удобным «классическим» методом N-алкилирования азотсодержащих гетероциклических соединений является их взаимодействие с соответствующим алкилирующим агентом в среде безводного диметилформамида при наличии калия карбоната, при температуре 20-25 °С [8]. Этот метод широко используется в тех случаях, когда N-алкилирование не сопровождается образованием изомерных продуктов. Авторами обнаружено, что N-алкилирование бензимидазола, 2-оксибензимидазола и бензотриазола бензилбромацетатом в указанных условиях приводит исключительно к N1-замещенным производным, а в случае аденина единственным направлением реакции является N9-замещение с удовлетворительным выходом целевых продуктов (36-48%) (рис. 2).
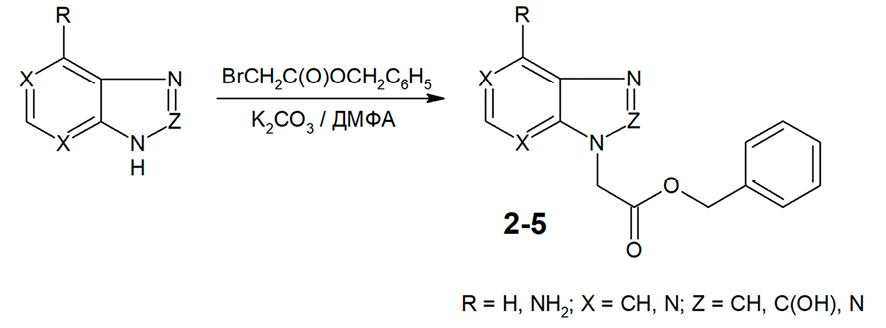
Рис. 2. Алкилирование бензимидазола, 2-оксибензимидазола, бензотриазола и аденина бензилбромацетатом Примечание: составлено авторами на основе полученных данных в ходе исследования
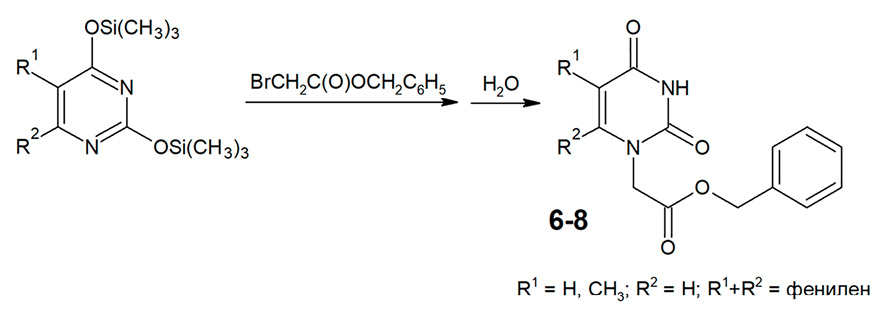
Рис. 3. Алкилирование триметилсилилпроизводных урацила, тимина и хиназолин-2,4(1Н,3Н)-диона бензилбромацетатом Примечание: составлено авторами на основе полученных данных в ходе исследования
Алкилирование урацила и его производных в идентичных условиях приводит к смеси N1-моно- и N1,N3-дизамещенных производных, разделение которых часто бывает малоэффективным. Особенно осложнено N-алкилирование производных урацила, содержащих заместители в положении 6 пиримидиновой системы, в том числе хиназолин-2,4(1Н,3Н)-диона, что приводит к стерическим затруднениям процессов алкилирования в положении N1 [9]. В связи с этим для повышения региоселективности N-алкилирования в среде диметилформамида при наличии калия карбоната используются иные субстраты, в частности N1,N3-дибензоилпроизводные пиримидиновых оснований [10; 11]. Также для селективного получения N1-монозамещенных производных урацила широко применяется модифицированный метод Гилберта – Джонсона, основанный на использовании триметилсилилпроизводных урацила и его аналогов в качестве субстратов [12]. Недостатком этого метода является то, что в нем могут быть использованы только алкилирующие агенты с высокой реакционной способностью, например альфа-галоидэфиры. Сложные эфиры бромуксусной кислоты обладают значительно более низкой реакционной способностью по сравнению с альфа-галоидэфирами, в связи с чем их применение в обычных условиях этого метода (неполярная или малополярная среда, комнатная температура) не приводит к ожидаемым результатам. Ранее авторами была разработана оригинальная модификация метода Гилберта – Джонсона, существенно расширившая ряд используемых алкилирующих агентов. Сущность этого метода «сплавления» заключается во взаимодействии триметилсилилпроизводных урацила с такими алкилирующими агентами, обладающими средней реакционной способностью, как 1-бром-2-феноксиэтаны или сложные эфиры альфа-галогенкарбоновых кислот, при нагревании без растворителя при температуре 140-180 °С с удалением образующегося триметилхлорсилана или триметилбромсилана в вакууме [13]. В настоящем исследовании авторы использовали триметилсилилпроизводные урацила, тимина и хиназолин-2,4(1Н,3Н)-диона, выход целевых продуктов составил 50-68% (рис. 3).
Экспериментальная часть
Спектры ЯМР полученных соединений регистрировали на спектрометре Bruker Avance 600 (600 МГц для 1H и 150 МГц для 13C) в ДМСО-d6, внутренний стандарт – тетраметилсилан. Тонкослойная хроматография выполнена на пластинах TLC Silica gel 60 F254 с последующим проявлением в УФ-свете. Определение температуры плавления проводили в стеклянных капиллярах с помощью прибора Mel-Temp 3.0 (Laboratory Devices Inc., США).
Синтез N-замещенных сложноэфирных производных азотсодержащих гетероциклических соединений «классическим» методом
Соответствующий азотсодержащий гетероцикл (0,016 моль) и 3,5 г (0,024 моль) тонко измельченного калия карбоната перемешивают 30 мин. в 40 мл безводного ДМФА при комнатной температуре, добавляют 3,0 г (0,018 моль) бензилбромацетата и перемешивают при той же температуре в течение 48 ч. Фильтруют, фильтрат упаривают в вакууме, остаток растирают с 50 мл воды и выдерживают при температуре 0-5 °С в течение суток. Образовавшийся осадок отфильтровывают, промывают водой, сушат на воздухе и кристаллизуют из подходящего растворителя.
1-(2-Бензилокси-2-оксоэтил)бензимидазол (2). Выход 42%, Т. пл. 122-124 °С. Спектр ЯМР 1Н, δ, м. д.: 5,23 с (2Н, NСН2); 5,37 с (2Н, OСН2); 7,23-7,30 м (1Н, J = 7 Гц, Н5); 7,32-7,41 м (6Н, Н6, фенил); 7,57 д (1Н, J = 7 Гц, Н7); 7,72 д (1Н, J = 7 Гц, Н4); 8,27 с (1Н, Н2). Спектр ЯМР, 13С, δ, м. д.: 45,87; 67,07; 110,93; 119,94; 122,19; 123,01; 128,50; 128,71; 129,93; 134,77; 136,00; 143,68; 145,19; 168,77.
1-(2-Бензилокси-2-оксоэтил)-2-оксибензимидазол (3). Выход 36%, Т. пл. 106-110 °С. Спектр ЯМР 1Н, δ, м. д.: 4,84 с (2H, NCH2); 5,19 с (2H, OCH2); 7,07-7,09 м (2H, H5, H6); 7,19-7,21 м (2H, H4, H7); 7,32-7,37 м (5H, фенил). Спектр ЯМР 13С, δ, м. д.: 40,01; 42,51; 66,94; 108,87; 121,90; 128,39; 128,91; 129,38; 136,03; 153,92; 168,45.
1-(2-Бензилокси-2-оксоэтил)бензотриазол (4). Выход 40%, Т. пл. 105-109 °С. Спектр ЯМР 1H, δ, м. д.: 5,22 с (2H, NCH2); 5,88 с (2H, OCH2); 7,32-7,37 м (5H, фенил); 7,42 д (1H, J = 8 Гц, H4); 7,56 т (1H, J = 7,5 Гц, H6); 7.86 т (1H, J = 7,5 Гц, H5); 8,07 д (1H, J = 8 Гц, H7). Спектр ЯМР 13С, δ, м. д.: 40,01; 49,06; 67,32; 111,20; 119,63; 124,57; 128,06; 128,50; 128,92; 133,99; 135,81; 145,56; 167,74.
9-(2-Бензилокси-2-оксоэтил)аденин (5). Выход 48%, Т. пл. 218-219 °С. Спектр ЯМР 1Н, δ, м. д. :5,17 с (2Н, NСН2); 5,21 с (2Н, OСН2); 7,30-7,40 м (7Н, NH2, фенил); 8,16 с (1Н, Н8), 8,17 с (1Н, Н2). Спектр ЯМР, 13С, δ, м. д.: 43,96; 66,63; 118,27; 127,98; 128,21; 128,42; 135,39; 141,25; 149,73; 153,66; 155,97; 167,89.
Синтез N-замещенных сложноэфирных производных азотсодержащих гетероциклических соединений методом «сплавления»
Соответствующий азотсодержащий гетероцикл (0,016 моль) и 0,05 г аммония хлорида кипятят в 50 мл гексаметилдисилазана до образования прозрачного раствора. В ряде случаев, например при силилировании хиназолин-2,4(1Н,3Н)-диона, реакция силилирования протекает медленно и требует кипячения исходных реагентов в течение многих суток. Добавление 10 мл O,N-бис(триметилсилил)ацетамида, как более эффективного силилирующего агента, позволяет сократить длительность этого процесса до нескольких часов. Избыток гексаметилдисилазана удаляют в вакууме, к остатку добавляют 3,0 г (0,018 моль) бензилбромацетата и нагревают при температуре 160-165 °С в течение 2 ч при остаточном давлении 40-50 мм рт. ст. Охлаждают, добавляют 25 мл 95% этанола и 2 мл концентрированного аммония гидроксида и выдерживают при комнатной температуре в течение суток. Образовавшийся осадок отфильтровывают, промывают водой, сушат на воздухе и кристаллизуют из подходящего растворителя.
1-(2-Бензилокси-2-оксоэтил)урацил (6). Выход 56%, Т. пл. 196-200 °С. Спектр ЯМР-спектр, 1H, δ, м. д.: 4,59 с (2H, NCH2). 5,19 с (2H, OCH2); 5,61 д (1H, J = 8, H5); 7,32-7,39 м (5H, фенил); 7.63 д (1H, J = 8, H6);11,40 с (1H, NH). Спектр ЯМР 13C, δ, м. д.: 39,98; 49,13; 66,94; 101,61; 128,40; 128,93; 135,97; 146,28; 151,46; 164,19; 168,56.
1-(2-Бензилокси-2-оксоэтил)тимин (7). Выход 50%, Т. пл. 198-200 °С. Спектр ЯМР 1H, δ, м. д.: 1,75 с (3H, CH3); 4,55 с (2H, NCH2); 5,18 с (2H, OCH2); 7,33-7,37 м (5H, фенил); 7,51 с (1H, H6); 11,39 с (1H, NH). Спектр ЯМР 13C, δ, м. д.: 12,33; 39,99; 48,94; 66,89; 109,12; 128,39; 128,92; 136,00; 141,98; 151,44; 164,75; 168,65.
Расчетный спектр фармакологической активности 1-(2-бензилокси-2-оксоэтил)бензотриазола
|
Pa |
Pi |
Активность |
|
0,764 |
0,010 |
Ингибитор АДФ-тимидинкиназы |
|
0,729 |
0,022 |
Ингибитор НАДФ-пероксидазы |
|
0,724 |
0,029 |
Ингибитор проницаемости мембран |
|
0,709 |
0,033 |
Ингибитор D-глюкозилфосфадитолинозинфосфолипазы |
|
0,681 |
0,018 |
Ингибитор фталат-4,5-диоксигеназы |
|
0,655 |
0,012 |
Ингибитор митохондриальной пептидазы |
|
0,665 |
0,024 |
Ингибитор эндо-1,6-бета-глюкозидазы |
|
0,651 |
0,029 |
Ингибитор сфинганинкиназы |
|
0,618 |
0,018 |
Ингибитор креатининазы |
Примечание: Pa – вероятность (0-1) наличия активности; Pi – вероятность (0-1) отсутствия активности.
Составлено авторами на основе полученных данных в ходе исследования.
1-(2-Бензилокси-2-оксоэтил)хиназолин-2,4(1Н,3Н)-дион (8). Выход 68%, Т. пл. 233-236 °С. Спектр ЯМР 1Н, δ, м. д.: 5,00 с (2H, NCH2); 5,20 с (2H, OCH2); 7,29 т (1H, J = 8 Гц, H6); 7,34-7,37 м (6H, фенил, H8); 7,71 т (1H, J = 8 Гц, H7); 8,03 д (1H, J = 8 Гц, H5); 11,76 с (1H, NH). Спектр ЯМР 13С, δ, м. д.: 43,75; 66,51; 114,51; 115,43; 122,91; 127,60; 127,85; 128,13; 128,39; 135,31; 135,53; 140,83; 150,29; 161,63; 168,14.
Вычислительный прогноз фармакологической активности новых соединений был выполнен в ИИ-среде PASS Online [14]. Наиболее выраженной и таргетированной активностью среди всех синтезированных соединений, согласно прогнозу in silico, обладает 1-(2-бензилокси-2-оксоэтил)бензотриазол (4) (таблица). В таблице представлены прогнозируемые виды активности соединения 4, для которых разница между наличием и отсутствием вероятности активности составляет не менее 0,6.
Результаты прогноза свидетельствуют о том, что ведущим в спектре фармакологической активности новых соединений является ингибирование различных оксиредуктаз, что косвенно свидетельствует о вероятном наличии у них противоопухолевой активности [15]. Об этом же свидетельствует и потенциальная способность полученных веществ подавлять важнейшие пути клеточного метаболизма. Образцы всех синтезированных соединений переданы в Лабораторию клеточных технологий Научного центра инновационных лекарственных средств ВолгГМУ для тестирования in vitro.
Заключение
Синтезирован ряд соединений – сложноэфирных производных азотсодержащих гетероциклов, представляющих интерес в качестве потенциальных противоопухолевых лекарственных средств.
Conflict of interest
Financing
Библиографическая ссылка
Тимофеева Ю.А., Покровская Ю.С., Финагеева М.О., Кузьминов О.В. СИНТЕЗ И ИССЛЕДОВАНИЕ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТИ IN SILICO НОВЫХ N-(2-БЕНЗИЛОКСИ-2-ОКСОЭТИЛ) ПРОИЗВОДНЫХ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ // Международный журнал прикладных и фундаментальных исследований. 2025. № 12. С. 42-47;URL: https://applied-research.ru/en/article/view?id=13779 (дата обращения: 01.07.2026).
DOI: https://doi.org/10.17513/mjpfi.13779