
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
STRUCTURAL GENOME VARAITIONS MANIFESTED AS A DUPLICATION OF THE CHROMOSOME 5 SHORT ARM ASSOCIATED WITH MOSAIC ANEUPLOIDY

Введение
В течение последнего десятилетия, молекулярная цитогенетика сделала значительные шаги вперед, направленные на разработку технологий, которые способны обнаруживать геномные перестройки с недоступным ранее разрешением [1-4, 8-13]. В результате был обнаружен широкий спектр ранее неизвестных микроделеционных и микродупликационных синдромов [2, 4, 8, 12, 13]. Кроме того, были описаны многочисленные уникальные хромосомные (субхромосомные) аномалии, дающие значимую информацию относительно происхождения и последствий геномных перестроек [8, 9, 12]. Известно, что отдельные генные мутации, вариации числа копий ДНК (делеции/дупликации, анеуплоидии, транслокации) могут стать причиной геномной или хромосомной нестабильности (CIN), проявляющейся в виде структурных или численных изменений хромосом [1, 6, 7, 15]. В этом контексте описание случаев, в которых наблюдаются регулярные (немозаичные) генетические изменения и повышенный уровень соматических мутаций или CIN, имеют принципиальное значение [6, 15]. Сочетание молекулярно-цитогенетических методов исследования всего генома и отдельных клеток с биоинформатическим методом дает возможность связать геномные изменения с конкретной молекулярной или клеточной патологией [7].
Цель работы
Целью работы явилось исследование геномных аномалий у ребенка с умственной отсталостью, врожденными пороками развития и повышенным уровнем спорадической анеуплоидии (CIN) с помощью современных молекулярно-генетических методов сканирования генома.
Материалы и методы исследования
В настоящей работе было проведено цитогенетическое и молекулярно-цитогенетическое исследование клеточного материала мальчика в возрасте 1 года 3 месяцев с множественными пороками развития. Препараты метафазных хромосом получали из лимфоцитов периферической крови, культивируемых in vitro, стандартным методом. Цитогенетический анализ проводили на хромосомных препаратах с использованием светового микроскопа при увеличении х1125. Хромосомы идентифицировали при помощи метода дифференциального окрашивания хромосом по длине (G- и C-методы), который осуществлялся по общепринятому протоколу [1, 2]. Для определения частоты CIN, было проведено исследование методом флюоресцентной гибридизация in situ (FISH) с ДНК зондами на хромосомы 1, 7, 16, 17, 18, X и Y, как описано ранее [16]. Высокоразрешающее полногеномное сканирование при помощи SNP array проводили на ДНК, выделенной из лимфоцитов периферической крови с использованием системы платформы фирмы Affymetrix, Santa и чипов, состоящих из ~ 2,7 млн маркеров для оценки CNV и ~ 750000 для оценки SNP и программного обеспечения Affymetrix Chromosome Analysis Suite, описанного ранее [5,7]. Геномная локализация и гены, расположенные в перестроенных участках, были определены с помощью системы NCBI Build GRCh37/hg19 с эталонной последовательностью ДНК. Вариации числа копий исследовались с помощью биоинформатического анализа, описанного ранее [7].
Результаты исследования и обсуждения
Пациент исследовался в возрасте 1 года 3 месяцев из-за отставания в психомоторном развитии, нарушения зрения, снижения аппетита, срыгивания и малой прибавки в весе. Ребенок родился от первой беременности путем экстренного кесарева сечения в 34 недели в связи с признаками фетоплацентарной недостаточности плода. Масса тела при рождении была 1297г, длина – 38см; оценка по шкале Апгар составила 1/4 балла. При рождении отмечалась геморрагическая сыпь, тяжелая дыхательная недостаточность. На нейросонографии определялись признаки поликистозной дегенерации головного мозга. Мальчик с рождения находился на искусственном вскармливании, плохо прибавлял в весе, длительно сохранялись срыгивания пищей после кормления. При проведении компьютерной аудиометрии в возрасте 1 года выявлены признаки нейро-сенсорной двусторонней тугоухости II-III степени, при проведении ЭЭГ выявлена фокальная эпилептиформная активность в затылочно-теменно-височной области слева.
При обследовании мальчика в возрасте в 1 года 3 месяцев обращали на себя внимание следующие признаки: значительный дефицит массы тела и окружности головы (менее 3-й центили), множественные лицевые микроаномалии: долихомикроцефалия, высокий лоб, гипотелоризм глазных щелей, эпикант, длинный фильтр, низкопосаженные ушные раковины, гипоплазия эмали зубов. В неврологическом статусе отмечен левосторонний спастический гемипарез легкой степени, при этом тонус мышц в правых конечностях был снижен. Выявлено значительное отставание в психомоторном развитии: по шкале CAT/CLAMS коэффициент развития составлял менее 50%, что соответствует грубой задержке развития; при осмотре ребенка обращали на себя внимание постоянные стереотипные движения в виде постукивания рукой по голове; отсутствовала реакция на обращенную речь, не было интереса к игрушкам; речь представляла спонтанное монотонное гуление. Выявлены также атрофия зрительных нервов нисходящего генеза; сходящееся косоглазие, нистагм, катаракта слева; при проведении МРТ головного мозга обнаружена врожденная аномалия развития в виде гипоплазии мозолистого тела.
При проведении цитогенетического анализа у ребенка было обнаружено наличие CIN в виде анеуплоидии. На рис. 1 представлены результаты цитогенетического исследования мальчика. Кариотипы родителей были нормальными.
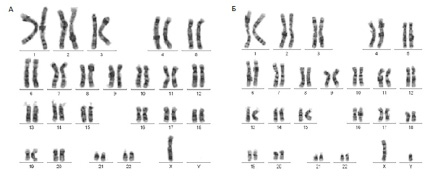
Рис. 1. Результаты цитогенетического исследования методом G – окрашивания (А – метафаза без хромосомы Y, Б – нормальны кариотип)
Для определения частоты CIN было проведено исследование методом флюоресцентной гибридизация in situ (FISH) с ДНК зондами на хромосомы 1, 7, 16, 17, 18, X и Y. Анализ FISH подтвердил наличие CIN (анеуплоидии) и показал, что частота соматических/спорадических хромосомных мутаций у пациента в 3-7 раз выше по сравнению с нормой, представленной в предыдущих исследованиях [1, 2, 6, 10, 15, 16].
Методом высокоразрешающего полногеномного сканирование при помощи SNP array была выявлена дупликация, расположенная в участке 5p13.3p13.2 (размером ~ 994 тыс. пн), она охватывала 11 генов, функции шести из которых известны и они индексированы в базе данных OMIM (Online Mendelian Inheritance in Men) (http://omim.org/) (рис. 2). Биоинформатический анализ показал, что эта дупликация, вероятно, является причинной задержки развития и врожденных пороков развития, наблюдаемых у пациента. Кроме того, это позволило нам определить молекулярный механизм геномной нестабильности, приводящей к развитию выявленной патологии центральной нервной системы.

Рис. 2. Схематическое изображение дупликации участка короткого плеча хромосомы 5, обнаруженной с помощью молекулярного кариотипирования с помощью геномного браузера UCSC (http://genome.ucsc.edu/)
В течение последних десятилетий в литературе было несколько сообщений о дупликации в участке 5p13. Молекулярно-цитогенетические исследования свидетельствуют, о том, что подобные дупликации вызывают синдром дупликации 5p13 (OMIM:613174) с участием гена NIPBL. Клинически данный синдром характеризуется задержкой развития и умственной отсталостью со следующими микроаномалиями лица: выступающие лобные бугры, большой или широкий лоб, короткие/наклонных глазные щели, высокое нёбо, и низкорасположенные ушные раковины [14]. Тем не менее, в данном случае дупликация не затронула критический участок синдрома дупликации 5p13. В базе данных DECIPHER v8.8 (https://decipher.sanger.ac.uk) было описано два случая с дупликацией, аналогичной перестройке у нашего пациента. Хотя у одного из пациентов отмечалась грубая задержка развития, в обоих случаях, похоже, наблюдались более легкие фенотипические появления по сравнению с описываемым нами пациентом. Фенотипические различия, вероятно, объясняются вариациями размеров исследуемого участка последовательностей ДНК. Для анализа корреляции генотип-фенотип мы использовали биоинформатический анализ [7].
При проведении микроматричного анализа (рис. 2) было выявлено шесть генов, которые были исследованы с помощью биоинформатического метода (для подробного описания использованных баз данных см. [7]). Ген TARS (OMIM:187790) кодирует различные формы аминоацил-тРНК-синтетазы и взаимодействует с многочисленными белками этого семейства. Показано, что другие функции при нарушении гена TARS могут быть связаны с расстройствами нервной системы и аутоиммунными заболеваниями. Ген ADAMTS12 (OMIM:606184) кодирует дезинтегрин и металлопротеиназу из семейства белков протромбоспондина, связанных с защитной от опухоли функцией, взаимодействуя с рядом генных продуктов, среди которых белки, вовлеченные в геномную сеть Notch (NOTCH1, FURIN). Функциональное изменение числа копий гена ADAMTS12 может быть связано с многочисленными патогенными процессами, включая те, которые ассоциированы с дисфункцией головного мозга, аутоиммунными, воспалительными и онкологическими заболеваниями. Ген RXFP3 (OMIM:609445) является частью семейства релаксин пептидных рецепторов. Биоинформатический анализ показал, что вариации числа копий гена RXFP3 может быть причиной изменения нейропептидов, организации нейроэндокринных сигналов и стимулирования усваивания воды и пищи. Ген SLC45A2 (OMIM:606202) кодирует белок, являющийся промежуточным звеном синтеза меланина, и он связан с альбинизмом IV типа (OMIM:606574). С помощью BioGPS было установлено, что этот ген активнее экспрессируется в сетчатке (http://biogps.org/#goto=genereport&id=51151). Ген AMACR (OMIM:604489) кодирует рацемозу и, вероятно, мутации гена во взрослом возрасте могут вызвать сенсомоторную нейропатию, пигментную ретинопатию, и адреномиелонейропатию из-за дефектов в синтезе желчных кислот. Кроме того, продукт этого гена взаимодействует с многофункциональными белками, которые играют роль в транскрипции и регуляции клеточного цикла, запрограммированной гибели клеток и поддержании стабильности генома. Ген C1QTNF3 (OMIM:612045) кодирует C1q и белок фактора некроза опухоли, мутации в котором связаны с заболеваниями нервной системы, приводящими к нарушению поведения. Продукт гена взаимодействует с лептином, который является элементом геномной сети, регулирующей размер тела, жировой запас, и может воздействовать на мозг, ингибируя потребление пищи.
Биоинформатический анализ генов, затронутых в дупликации, позволил нам предположить корреляцию генотип-фенотип. Таким образом, увеличение числа копий гена TARS, скорее всего, будет связано с задержкой развития и врожденными пороками развития; гена ADAMTS12 – с мозговой дисфункцией, а также с задержкой развития и врожденными пороками развития; гена RXFP3 – с неврологическими симптомами и снижением аппетита; гена SLC45A2 – с нарушением зрения; гена AMACR – с неврологическими симптомами, включая глухоту, нарушение зрения и другими врожденными пороками развития; гена C1QTNF3 – с задержкой роста и снижением аппетита.
Заключение
В настоящей работе дается описание случая дупликации короткого плеча хромосомы 5 в участке 5p13.3p13.2, ассоциированной с задержкой развития, врожденными пороками развития и CIN (спорадической анеуплоидией). Применение высокоразрешающего SNP микроматричного анализа, FISH исследования соматической анеуплоидии и биоинформатического метода исследования позволили провести корреляцию генотип-фенотип и выявить связь между регулярной дупликацией 5p13 и CIN (т.е. спорадической мозаичной анеуплоидией низкого уровня) у представляемого пациента. Таким образом, совместное использование методов, направленных на исследование индивидуальных и межклеточных вариации генома в сочетание с углублённым биоинформатическим анализом, может выявить патогенное значение этих вариаций и понять основную причину фенотипических вариаций геномных перестроек, связанных с механизмом заболевания в каждом конкретном случае (персонифицированная геномика).
Исследование выполнено за счет гранта Российского Научного Фонда (проект №14-15-00411).
Библиографическая ссылка
Ворсанова С.Г., Юров Ю.Б., Куринная О.С., Алямовская Г.А., Кешишян Е.С., Демидова И.А., Юров И.Ю. СТРУКТУРНАЯ ВАРИАЦИЯ ГЕНОМА В ВИДЕ МИКРОДУПЛИКАЦИИ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 5, АССОЦИИРОВАННАЯ С МОЗАИЧНОЙ АНЕУПЛОИДИЕЙ // Международный журнал прикладных и фундаментальных исследований. 2015. № 9-1. С. 39-43;URL: https://applied-research.ru/en/article/view?id=7435 (дата обращения: 01.07.2026).