
Scientific journal
International Journal of Applied and fundamental research
ISSN 1996-3955
ИФ РИНЦ = 0,556
PRACTICAL SYNTHESIS OF BETULIN DIACETATE AND BETULIN SUCCINATE AS POTENTIAL LIPID-LOWERING SUBSTANCES

Бетулин – компонент бересты березы, по своим свойствам и строению близок к липофильным стеролам, которые относительно легко проникают через плазматические мембраны, а затем свободно секретируются клетками стероидогенных эндокринных желез. [1] Известно, что для всех производных бетулина (бетулиновой и бетулоновой кислот, сукцинатов, ацетатов и других эфиров органических кислот) характерны близкие фармакологические эффекты – противоопухолевые, гиполипидемические, гепатопротекторные и противовирусные, проявляющиеся в различной степени в зависимости от структуры [2-4]. Примером этого является работа F.B. Mullauer et al. по сопоставлению влияния бетулина и бетулиновой кислоты на противоопухолевую активность. Показано, что бетулиновая кислота, как противоопухолевый агент, существенно уступает своему более липофильному предшественнику – бетулину с двумя спиртовыми группами, за счет совместного действия комплексов или ассоциатов бетулина с холестеролом. [5] Взаимодействие бетулина с холестеролом, вероятно, близко к доказанной ассоциации β-ситостерола с холестеролом [6], которая в конечном счете приводит к гиполипидемическому эффекту, поскольку для достижения последнего немаловажным является большая липофильность тритерпеноида. Этот факт нашел отражение в работах Василенко и др. по экспериментальному доказательству гиполипидемических и гепатопротекторных свойств более липофильного, чем бетулин тритерпенового соединения – диацетата бетулина [7].
Однако, несмотря на многочисленные публикации, посвященные исследованию фармакологического действия производных бетулиновой кислоты, гиполипидемических лекарственных средств на основе эфиров бетулина на фармацевтическом рынке не представлено. Это связано, главным образом, с отсутствием надежных препаративных методов синтеза эфиров бетулина как потенциальных лекарственных веществ, требующих достижения высокой чистоты целевого продукта, отсутствие токсичных реагентов и прекурсоров в процессе его получения, технологичность, экологичность и экономичность синтеза.
В настоящей работе нами экспериментально и теоретически оценены известные методы синтеза диацетата и сукцинатов бетулина, для которых экспериментально на крысах доказаны гиполипидемический и гепатопротекторный эффекты, и даны рекомендации для препаративных методик их получения.
Материалы и методы исследования
Реактивы. Бетулин (C30H50O2) получали в соответствии со способом, указанном в литературе [8], tпл. 260 °C (лит. 254–256 °C); чистота 99.5 %, ИК, ν, cm−1: 3470 st (OH), 1640 st (C=C); 1H-ЯМР (CDCl3) (δC, м.д.): 4.67 m (1H, =CH2), 4.57 m (1H, =CH2), 3.78 br. s (1H, 28-CH2OH), 3.31 m (1H, 28-CH2OH), 3.17 m (1H,3-CHOH), 2.36 m (1H, 19-CH), 1.66 s (3H, CH3), 1.23 s (3H, CH3), 0.96 s (3H, CH3), 0.94 s (3H, CH3), 0.80 s (3H, CH3), 0.74 s (3H, CH3). 13C-ЯМР (CDCl3) (δC, м.д.): 76.71 (C-3), 109.46 (C-29), 150.24 (C-20), 57.87 (C-28). EI-MS m/z ( %): 442 (M+, 40), 411 (60), 203 (95), 189 (100), 95 (85). Вода деионизованная (сопротивление>18 MΩ cm, Simplicity, Millipore Inc.) с рН 5.5 при температуре 20 ± 1 °C, янтарный ангидрид (ТУ 6-09-3611-85), ледяная уксусная кислота (ГОСТ 81-75 изм.№3), имидазол (ТУ 6-09-37-1127-91), хлористоводородная кислота (ГОСТ 3118-77), ортофосфорная кислота 85 % (ГОСТ 6552-80), метилен хлористый (ТУ 2631-019-44493179-98), изопропиловый спирт (ГОСТ 9805-84), этанол (Гост 5964-93), ацетон (ГОСТ 2603-79), ацетонитрил (ТУ 6-09-06-1092-83), этилацетат (ГОСТ 22300-76), пара-толуолсульфокислота (п-ТСК) (ТУ 6-09-3668-77) использовались без предварительной очистки и какой-либо обработки.
Приборы и оборудование. ИК – спектры были получены на приборе Shimadzu IR-Prestige-21 (KBr табл.). ОФ-ВЭЖ – хроматограммы эфиров бетулина были получены на высокоэффективном жидкостном хроматографе марки Shimadzu LC-20 Avp, колонка Discovery C18 (250 × 4.6 mm, 5μm) с UV-детектором. 13С, 1Н ЯМР спектры регистрировали на спектрометрах «Bruker Advance DPX – 200» и «Bruker DRX SF = 500» в CDCl3, внутренний стандарт - ТМС.
Методика синтеза диацетата бетулина по А.Н. Кислицыну [9] В реактор загружали бетулин (3,0 г, 6,78 ммоль), этилацетат (16,2 г, 0,184 моль), уксусную кислоту (16,2 г, 0,270 моль) и 150 мг п-ТСК. Исходную смесь нагревали до температуры кипения. Образующиеся пары после конденсации в дефлегматоре поступали в устройство, предназначенное для избирательного удаления воды с использованием адсорбента – силикагеля. Обезвоженный дистиллят возвращали в реактор. Процесс проводили в условиях кипения в течение 4 ч. Затем реакционную смесь упаривали, охлаждали до 5-10 °C для кристаллизации диацетата бетулина. После перекристаллизации продукта из изопропанола (массовое соотношение диацетат:спирт – 1:10) при 20-25 °C получали 3,29 г диацетата бетулина (выход 92 ± 2 % (n = 5)). ИК-спектр (KBr) (ν, cм-1): 3068,75 (С=С); 2947,23; 2870,08 (С-Н); 1737,86 (С=О); 1456,26; 1388,75; 1365,60 (С-С); 1244,09; 1031,92 (С-О-С), 979,84 (С-О), 889,18 (С=С); 1H-ЯМР (CDCl3) (δC, м.д.): 4,72 (1H, м, =CH2); 4,55 (1H, м, =CH2); 4,47 (1H, м, H3); 4,26 (1H, д, J10.7 Гц, Н28); 3,86 (1Н, д, J10.7 Гц, Н28); 2,50 (1Н, м, Н19); 2,08(3Н, с, CH3CO); 2,03 (3H, c, CH3CO); 1,65; 1,02; 0,94; 0,82; 0,80 (все 3H, c, CH3); 13С-ЯМР (CDCl3) (δC, м.д.): 14,95 (С27), 15,79 (С24), 15,92 (С25), 16,27 (С26), 17,94(С6), 18,89 (С29), 20,58 (С11), 20,77 (СН3Ас), 21,04(СН3Ас), 23,46 (С2), 24,92 (С12), 26,83 (С15), 27,71(С23), 29,35 (С16), 29,52 (С21), 33,91 (С7), 34,32(С22), 36,82 (С10), 37,32 (С13), 37,55 (С4), 38,16(С1), 40,66 (С8), 42,45 (С14), 46,08 (С17), 47,47(С19), 48,54 (С18), 50,05 (С9), 55,15 (С5), 62,50(С28), 80,61 (С3), 109,69 (С30), 149,80 (С20), 170,63(СОАс), 171,23 (СОАс).
Методика синтеза диацетата бетулина по В.А. Левданскому [10] В колбу объемом 250 мл, снабженную обратным холодильником, загружали 4,42 г (0,01 моль) бетулина, заливали 50 мл ледяной уксусной кислоты и добавляли 30 г H3PO4 85 %. Смесь кипятили на воздушной бане в течение 1,5 ч, затем реакционную массу выливали в стакан объемом 1 л с 250 мл холодной воды. Выпавший продукт отфильтровывали и промывали на фильтре водой до нейтральных рН промывных вод. Далее осадок высушивали. Выход диацетата бетулинола составил 3,42 г (65 ± 3 %) (n = 5).
Методика синтеза моно- и дисукцинатов бетулина по П.А. Красутскому [11] а) Сукцинат бетулина В колбу объемом 25 мл помещали 1,00 г бетулина (2,26 ммоль), 0,249 г янтарного ангидрида (2,49 ммоль) и 0,462 г имидазола (6,79 ммоль). Добавляли 20 мл сухого дихлорметана, перемешивали в течение 24 часов. После окончания реакции добавляли 10 мл 3 % HCl до рН=2,0 при медленном перемешивании. Отделяли органический слой и объединяли с дихлорметановыми экстрактами водного слоя (3х5 мл), затем промывали 3 % HCl (2х10 мл) и сушили безводным Na2SO4. После удаления растворителя получали 0,78 г (1,44 ммоль) белого порошка, в который добавляли 1 мл ацетона. После сушки получали 0,60 г белого порошка, содержащего смесь моно- и дисукцинатов в молярном соотношении 60:40. Выход моносукцината после перекристаллизации составил 49 ± 2 %.
ИК (KBr), ν, cm−1: 3361.93, 2941.44, 1732.08, 1263.37, 1172.72 см-1; 1H-ЯМР (CDCl3) (δC, м.д.): 4.67 (1H, с, CH2=C), 4.30 (1H, д, J=11.1 Гц), 2,66(1H, д, J=11.1 Гц), 2,65 (1Н, м), 1.67 (3H, с), 0.75, 0.81, 0.96, 1.02 (4х3H, все с), 0.71-2.1 (комплекс, 28Н); 13С-ЯМР (CDCl3) (δC, м.д.): 177.26, 172.47, 150.06, 109.84, 79.06, 77.32, 77.01, 76.69, 63.14, 60.50, 55.26, 50.36, 48.78, 47.75, 46.40, 42.68, 40.84, 38.81, 37.58, 34.14, 29.71, 28.94, 27.95, 25.18, 20.81, 19.07, 18.28, 16.49, 16.00, 15.36, 14.76, 14.74.
Сырец моносукцината бетулина ИК (KBr), ν, cm−1: 3361.93, 2941.44, 1732.08, 1712.79, 1280.73, 1263.37, 1172.72, 1161.15 см-1; 1H-ЯМР (CDCl3) (δC, м.д.): 4.67, 4.58, 4.30, 4.28, 2.67, 2.66, 2.65, 2.64, 1.67, 1.63, 0.75, 0.81, 0.96, 1.02, 0.71-2.1 (комплекс, 28Н); 13С-ЯМР (CDCl3) (δC, м.д.): 177.26, 177.10, 172.47, 172.17, 150.42, 150.06, 109.84, 109.67, 79.06, 79.05, 77.32, 77.01, 76.69, 63.14, 60.80, 60.50, 55.26, 55.25, 50.36, 50.34, 48.78, 48.73, 47.75, 47.72, 46.40, 42.68, 42.67, 40.84, 38.81, 38.67, 37.58, 37.28, 34.14, 33.95, 29.71, 29.69, 29.08, 28.94, 27.95, 27.26, 25.18, 20.81, 20.76, 19.13, 19.07, 18.28, 18.13, 16.49, 16.12, 16.08, 16.00, 15.96, 15.36, 14.76, 14.74.
б) Дисукцинат бетулина В колбу объемом 25 мл помещали 0,5 г бетулина (1,13 ммоль), 0,34 г янтарного ангидрида (3,40 ммоль) и 0,46 г имидазола (6,76 ммоль). Добавляли 15 мл сухого дихлорметана, перемешивали в течение 12 часов. Добавляли 10 мл 3 % HCl, отделяли дихлорметановый органический слой, включая экстракты водного слоя дихлорметаном (3х5 мл), промывали 3 % HCl (2х10 мл), затем сушили Na2SO4. После удаления растворителя получали 0,71 г (1,10 ммоль) порошка, имеющего желтый тон. Перекристаллизация из смеси хлороформ-гексан давала 0,65 г (1,02 ммоль) порошка желтоватого оттенка. Выход моносукцината после перекристаллизации составил 90 ± 3 %. ИК (KBr), ν, cm−1: 2945.30, 1732.08, 1161.15 см-1; 1H-ЯМР (CDCl3) (δC, м.д.): 4.68 (1H, с, =CH2), 4.59 (1H, с, =CH2), 4.51 (1Н, м), 4.32 (1H, д, J=11.4 Гц, OCH2), 3.89 (1H, д, J=10.8 Гц, OCH2), 2.67(8Н, м, цепочка сукцината), 2.44 (1Н, м), 1.68 (3H, с), 1.03, 0.98, 0.97, 0.83, 0.76 (5х3H, все с), 1.03-2.1(24Н, комплекс); 13С-ЯМР (CDCl3) (δC, м.д.): 177.59, 177.50, 172.32, 171.73, 150.08, 109.86, 81.54, 63.18, 55.39, 50.24, 48.76, 47.71, 46.42, 42.68, 40.87, 38.34, 37.82, 37.58, 37.04, 34.38, 34.07, 29.68, 29.61, 29.53, 29.33, 29.10, 28.95, 27.87, 27.00, 20.80, 19.10, 18.13, 16.49, 16.13, 16.02, 15.35, 14.80.
Результаты исследования и их обсуждение
В табл. 1 приведены основные методики синтеза диацетата бетулина, различающиеся по степени экономичности и воспроизводимости результатов. Первую группу составляют методики получения непосредственно из бересты березы. Количественный результат и чистота целевого продукта в значительной степени будут зависеть от качества бересты (ареал произрастания, вид березы, сроки и условия хранения), а также методов очистки, что создает трудности для использования этих методов синтеза для получения фармацевтической субстанции, несмотря на привлекательность прямого получения продукта из исходного сырья.
В этом плане вторая группа методик синтеза диацетата бетулина этерификацией чистого бетулина является более подходящей для фармацевтической промышленности. Нами был выполнен синтез диацетата бетулина этерификацией уксусной кислотой в присутствии катализаторов (п-ТСК, либо H3PO4) по методикам А.Н. Кислицына [9] и В.А. Левданского [10], в которых не используется уксусный ангидрид, являющийся прекурсором (табл. 1).
Наибольшая чистота продукта с максимальным выходом достигалась синтезом по методике А.Н. Кислицына. [9] Общая схема реакции приведена на рис. 1.
Таблица 1
Основные методики синтеза диацетата бетулина
|
Источник |
Кислицын А.Н. [9] |
Левданский В.А. [10] |
Кузнецова С.А. [12] |
Сымон А.В. [13] |
Хлебникова Т.Б. [14] |
|
Реактивы |
Бетулин, ЭА*, CH3COOH, п-ТСК**, SiO2 |
Бетулин, CH3COOH, H3PO4 |
Береста, CH3COOH |
Бетулин, уксусный ангидрид |
Береста, CH3COOH**** |
|
Темпера-тура, °С |
Кип. |
Кип. |
Кип. |
Кип. |
Кип. |
|
Время, ч |
4-6 |
1-1,5 |
0,5-6 |
До полного растворения |
6-10 |
|
Выход, % |
90 (92±2, n=5***) |
95 (65±3, n=5***) |
47 % от массы абсолютно сухой бересты (перекрист. 89-95) |
64 |
38 % от массы абсолютно сухой бересты |
|
Примеси |
Бетулин 0,1 % моноацетат 2,9 % Неидентиф. вещества 6,9 % |
Бетулин, моноацетат, неидентиф. вещества |
Бетулин, моноацетат, неидентиф. вещества |
Бетулин, моноацетат, неидентиф. вещества |
Бетулин, моноацетат, неидентиф. вещества |
Примечания. * ЭА – этилацетат; **п-ТСК – паратолуолсульфокислота; ***наши данные по выходу диацетата; ****удаление воды в аппарате Сокслета.

Рис. 1. Получение диацетата бетулина по Кислицыну [9]
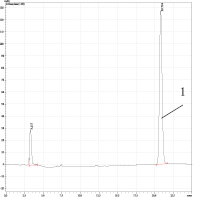
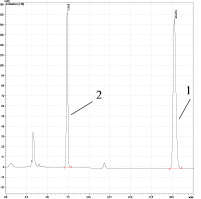
а) б)
Рис. 2. ОФ-ВЭЖХ диацетата бетулина. Условия хроматографирования: Колонка Discovery® C18, 25 cm x 4.6 mm, 5 μm (Supelco); объем пробы 20 мкл; скорость потока – 1,0 мл/мин; подвижная фаза – вода –ацетонитрил (v-v = 5-95); диоидно-матричный УФ детектор – λ=196 и 205 нм; температура – 40оС; время выхода – 20,79 мин. а) очищенный продукт синтеза по Кислицыну (пик 1); б) продукт синтеза по Кислицыну (пик 1) с введением в пробу бетулина (пик 2). Пик при τ=2,6 мин соответствует примеси в ацетонитриле
ОФ-ВЭЖ – хроматограмма целевого продукта, полученного по Кислицыну [9] и очищенного перекристаллизацией в изопропаноле приведена на рис. 2.
В качестве основных примесей по данным ОФ-ВЭЖХ-анализа выступали бетулин и моноацетат бетулина, концентрация которого по В.А. Левданскому достигала 40 % по отношению к диацетату. В качестве иллюстрации образования двух основных продуктов по Левданскому на рис. 3 приведены ИК-спектры перекристаллизованных продуктов реакции по сопоставляемым методикам (2 полосы для двух циклопентанпергидрофенантреновых циклов с ν 2943 и 2926 см-1).
Основные методики синтеза сукцинатов приведены в табл. 2.
Получение сукцинатов по методике С.А. Попова [16], в которой используется концентрат маточника после экстракции бересты березы, нами не рассматривалось по причине плохой воспроизводимости результатов и зависимости от качества используемого сырья.
Все методики предполагают использование в качестве этерифицирующего реагента янтарного ангидрида. В качестве мягкого акцептора протонов и компонента, структурирующего среду, использовали имидазол или пиридин, а в качестве среды использовали N-метилпирролидон, метилтретбутиловый эфир, либо пиридин при различных температурах (табл. 2). Нами была выбрана методика получения сукцинатов по Красутскому, в соответствии с которой синтез проводился в наиболее мягких условиях: комнатная температура и использование в качестве среды дихлорметана. Общая схема реакции приведена на рис. 4.
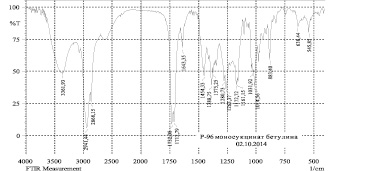
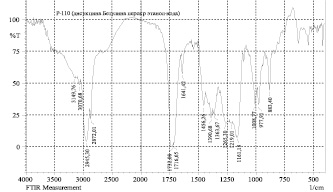
Нами показано по данным ЯМР-спектров, что синтез моносукцината бетулина протекал неселективно с образованием смеси моно- и дисукцината в молярном соотношении 3:2. В отличие от этого, дисукцинат образовывался с высоким выходом. На рис. 5 представлены ИК-спектры моно- и дисукцинатов. Поскольку как моно-, так и дисукцинат бетулина проявляют высокую гепатопротекторную активность [17], то можно предложить фармацевтическую субстанцию как в виде дисукцината, так и смеси сукцинатов.
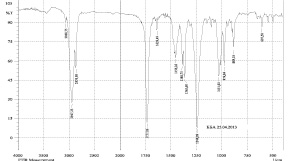
а)
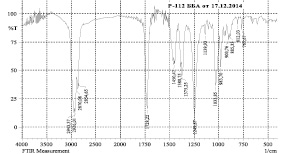
б)
Рис. 3. ИК-спектры диацетата, полученного по методу А.Н. Кислицина (а) и В.А. Левданского (б)
Таблица 2
Основные методики синтеза сукцинатов бетулина
|
Источник |
Глинский Я., [15] |
Попов С.А., [16] |
Красутский П.А. [11] |
Флехтер О.Б., [17] |
||
|
Реактивы |
Бетулин: имидазол: янтарный ангидрид = 1:4:1.08 |
Бетулин: имидазол: янтарный ангидрид = 1:4:4 |
Концентрат маточника: янтарный ангидрид: пиридин |
Бетулин: имидазол: янтарный ангидрид = 1:1,1:3 |
Бетулин: имидазол: янтарный ангидрид = 1:3,1:3 |
Бетулин: янтарный ангидрид |
|
Среда |
N-МПД* |
N-МПД* |
МТБЭ** |
CH2Cl2 |
CH2Cl2 |
пиридин |
|
Температура, °С |
Комн. |
70 |
30-35 |
Комн. |
Комн. |
Кип. |
|
Время, ч |
48 |
20 |
20 |
24 |
12 |
15 |
|
Выход, % |
92-95 |
73 |
95 |
73 (49 ± 2, n = 5****) |
82 (90 ± 3, n = 5****) |
92 |
|
Основной продукт |
28-моно-сукцинат |
3,28-ди-сукцинат |
28-моно-сукцинат |
28-моно-сукцинат |
3,28-ди-сукцинат |
3,28-ди-сукцинат |
|
Примеси*** |
Бетулин, дисукцинат |
Бетулин, моно-сукцинат |
Бетулин, дисукцинат |
Бетулин (0,1 %****), дисукцинат (30 %***) |
Бетулин (0,1 %****), моно-сукцинат (3-5 %****) |
Бетулин, моно- сукцинат |
Примечания. *N-МПД – N-метилпирролидон; **МТБЭ – метилтретбутиловый эфир; ***Количественный состав примесей не установлен; **** – наши данные по выходу продукта.
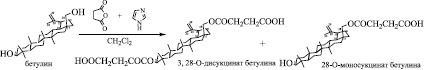
Рис. 4. Получение моно- и дисукцинатов бетулина по Красутскому
а)
б)
Рис. 5. ИК-спектры моносукцината (а) и дисукцината (б) бетулина
Таким образом, в работе нами оценены методы синтеза диацетата, моно- и дисукцинатов бетулина и предложены препаративные методики получения этих веществ, пригодные для фармацевтической промышленности. Показано, что оптимальной является методика получения диацетата бетулина ацетилированием уксусной кислотой, взамен токсичных уксусного ангидрида и пиридина, в среде этилацетата в присутствии пара-толуолсульфокислоты при избирательном удалении воды из реакционной смеси (использование ловушки для воды и адсорбента – силикагеля) при кипячении. Более перспективная методика получения дисукцината бетулина, позволяющей получить целевой продукт с более высоким выходом и чистотой, представляет собой этерификацию бетулина янтарным ангидридом в присутствии имидазола при комнатной температуре.
Библиографическая ссылка
Пегова Р.А., Гуленова М.В., Жильцова О.Е., Клабукова И.Н., Мельникова Н.Б. ПРЕПАРАТИВНЫЙ СИНТЕЗ ДИАЦЕТАТА И СУКЦИНАТОВ БЕТУЛИНА – ПОТЕНЦИАЛЬНЫХ ФАРМАЦЕВТИЧЕСКИХ СУБСТАНЦИЙ ГИПОЛИПИДЕМИЧЕСКОГО ДЕЙСТВИЯ // Международный журнал прикладных и фундаментальных исследований. 2015. № 10-2. С. 304-310;URL: https://applied-research.ru/en/article/view?id=7488 (дата обращения: 13.06.2026).