

Сахарный диабет 1 типа (СД1) – органоспецифическое аутоиммунное заболевание, которое развивается в результате T-клеточно-опосредованной деструкции инсулин-продуцирующих ß-клеток поджелудочной железы. Это аутоиммунное расстройство обусловлено как наследственными факторами, так и факторами окружающей среды [1, 2].
Многочисленные исследования позволили установить, что врожденные иммунные реакции, протекающие с участием Толл-подобных рецепторов (TLRs), могут способствовать развитию диабета у мышей. В частности, ß-клетки, вовлеченные в процесс апоптоза, могут активировать представляющие антиген врожденные иммуноциты через TLR2 [3]. В недавних исследованиях было показано, что панкреатические островки человека экспрессируют TLR2, TLR3, TLR4 и TLR9 [2].
Данные, полученные в экспериментах на животных моделях, а также людях показали, что системное воспаление играет важную роль в патофизиологических процессах развития диабета и способствует формированию врожденных иммунных реакций. TLRs являются ключевыми рецепторами врожденного иммунитета, которые распознают консервативные патоген-связанные молекулярные модели (PAMPs), вызывают воспалительные реакции, необходимые для защиты хозяина и инициируют адаптивный иммунный ответ.
Роль вирусной инфекции в инициации аутоиммунного воспаления в островковых клетках поджелудочной железы
Связь между СД1 и вирусной инфекцией впервые была установлена в 1864 году, когда клиническая манифестация СД1 совпала с заражением вирусом эпидемического паротита. В настоящее время известно, что РНК данного вируса проникает в островки Лангерганса как invitro, так и invivo. Спустя столетие, в 1969 году, была установлена связь дебюта СД1 и вируса краснухи. Эпидемиологические данные свидетельствуют о том, что связь вируса краснухи с развитием СД1 отмечается только у инфицированных пациентов, которые имеют генетическую предрасположенность к развитию СД1. Доказательства участия энтеровирусов в инициации СД1 были получены в нескольких исследованиях. При этом было отмечено увеличение концентрации энтеровирусной РНК в сыворотке крови по сравнению со здоровыми индивидуумами. В 1979 году была установлена связь цитомегаловирусной инфекции с развитием СД1. Тем не менее, аналогично эпидемическому паротиту и краснухе, энтеровирусная и цитомегаловирусная инфекции лишь повышают риск развития СД1 у генетически предрасположенных лиц.
Проникновение патогенов в организм человека контролируется врожденным и адаптивным звеньями иммунной системы. Адаптивный иммунитет опосредован В- и Т-лимфоцитами и распознает патогены посредством рецепторов с высокой аффинностью. Для формирования специфического иммунного ответа требуется несколько дней, поскольку реакции адаптивного иммунитета по нейтрализации микроорганизмов зависят от клеточной пролиферации, активации генов и синтеза белка. Более быстрые защитные механизмы обеспечиваются врожденным иммунитетом. В отличие от клоноспецифических рецепторов, которые экспрессируют Т- и В-лимфоциты, врожденная иммунная система использует неклональные наборы распознающих рецепторов, называемые паттерн-распознающими рецепторами (ПРР). Эти рецепторы связывают определенные молекулярные структуры, характерные для больших групп патогенов.
Существует несколько групп ПРР, которые могут секретироваться, экспресси-роваться на поверхности или внутри клетки. Наиболее важными представителями этого семейства являются TLRs.
TLRs представляют собой семейство распознающих рецепторов, которые играют важную роль в системе врожденного иммунитета путем активации провоспалительных сигнальных путей в ответ на внедрение патогенных микроорганизмов [4]. Комбинированная TLR4/моноцит и TLR3/ткань активация приводит к дальнейшему повышению уровня провоспалительных цитокинов. Следовательно, характер воспалительной реакции будет зависеть от степени экспрессии TLRs на лейкоцитах и клетках тканей, взаимодействия между лейкоцитами и клетками тканей, а также агонистов TLRs, с которыми они взаимодействуют [4].
Связывание PAMPsс TLRs индуцирует продукцию активных форм кислорода и азота, промежуточных соединений (ROI и РПП), провоспалительных цитокинов и до-регулирует экспрессию стимулирующих молекул, впоследствии инициирующих адаптивный иммунитет.
В настоящее время у человека идентифицировано 11 видов TLRs. По особенностям экспрессии TLRs можно разделить на универсальные (TLR1), с ограниченной экспрессией (TLR2, TLR4, TLR5) и специфические (TLR3). Наиболее распространенным рецептором является TLR1, что свидетельствует в пользу того, что этот рецептор участвует в регуляции передачи сигнала от других TLRs.
TLRs способны распознавать липиды, протеины, нуклеиновые кислоты как экзогенного происхождения (фрагменты микроорганизмов), так и образующиеся в организме.
Мутации генов TLRs и дефицит TLRs являются причиной повышенной восприимчивости организма к различным инфекциям и приводят к их хронизации [5].
Активация TLRs приводит в дальнейшем к активации клеток врожденной иммунной системы: макрофагов, дендритных клеток, тучных клеток, моноцитов, нейтрофилов, и индукции провоспалительных и противовоспалительных белков, белков острой фазы, костимулирующих молекул. Эти рецепторы участвуют также в формировании специфического иммунного ответа, влияя на механизмы «иммунологической памяти». Кроме того, TLRs играют важную роль в регуляции гемопоэза, влияют на дифференцировку миелоидных стволовых клеток, пролиферацию и дифференцировку Т- и В-лимфоцитов, активируют апоптоз. Активация TLRs, также как и рецептора ИЛ1, приводит к транскрипции ядерного фактора NF-kB через МАРК-киназы, что позволяет предположить, что эти рецепторы задействуют сходные сигнальные пути.
TLR2
Наиболее изученным TLR является TLR2. Он способен распознавать большой спектр основных компонентов клеточной стенки или РАМРs [6, 7]. Лигандами для TLR2 являются структуры грам-положительных бактерий, включая липопротеины, липопептиды, пептидогликаны и липотейхоевые кислоты, а также липоарабиноманнан микобактерии, грибковый зимозан, стафилококковый модулин и гликозилфосфатидилинозитол Tripanosomacruzi. Оставалось неясным, как один рецептор способен распознавать такой спектр структур. Позднее установили, что TLR2 формирует гетеродимерные комплексы с другими рецепторами. Димер TLR2/TLR6 распознает диацелированные липопептиды на моноцитах человека, а TLR1/TLR2 преимущественно триацелированные. TLR1-TLR2 гетеродимеризация активирует дендритные клетки, В-клетки, NK-клетки, тучные клетки и кератиноциты [1]. TLR2 и TLR6 участвуют в совместном выявлении зимозана дрожжевых грибков [1]. Распознавание грибковых паттернов TLR2, возможно, также осуществляет в комплексе с лектиновыми рецепторами. Кроме того, компоненты некротических, но не апоптотических клеток принимают участие в активации фибробластов и макрофагов с помощью TLR2 [8].
TLR4
TLR4 является одним из важнейших компонентов липополисахарид (ЛПС)-рецепторного комплекса, который активирует клетки при воздействии на грам-отрицательные бактерии. Тем не менее, он также взаимодействует с другими лигандами, в частности, растительного происхождения. Для распознавания ЛПС TLR4 требуется формирование белкового комплекса, содержащего дополнительные молекулы: ЛПС, как правило, связывается с ЛПС-связывающим белком сыворотки, и этот комплекс сначала распознается рецептором моноцитов и макрофагов, CD14, который, в свою очередь, взаимодействует с TLR4 и вызывает его активацию [9]. После связывания ЛПС с TLR4 и его ко-рецепторами – CD14 и MD-2, адаптерным белком MyD88 (миелоидный фактор дифференцировки 88) происходит соединение МДП (Toll/IL-1 рецептор) области TLR4 и MyD88. Это инициирует сигнальный каскад, который приводит к активации NF-kB пути, который, в свою очередь, активирует транскрипцию многих провоспалительных генов, кодирующих молекулы воспаления, включая цитокины, хемокины и другие эффекторы врожденного иммунного ответа [9].
Эндотоксический шок опосредован TLR4, который индуцирует высвобождение провоспалительных цитокинов и хемокинов из иммунных и неиммунных клеток. Например, отсутствие надлежащей TLR4-сигнализации может предрасполагать к сепсису у пациентов с ревматоидным артритом, получавших антагонист фактора некроза опухоли [10], но функциональный дефект TLR4 не предрасполагает к ревматоидному артриту сам по себе [11]. TLR4 требуется для поглотительной функции нейтрофилов при эндотоксин-индуцированном повреждении легких [12]. В 1998 году было установлено, что точечная мутация в гене, кодирующем TLR4, является молекулярной основой для гипер-чувствительности к липополисахариду у мышей линий C3H/HeJ и C57BL/10ScCr. Линия мышей C57BL/10ScCr имеет хромосомный дефект, который приводит к потере TLR4 гена [13]. Мыши, принадлежащие к этим линиям, показали фенотип мышей с делецией гена TLR4. У людей мутации TLR4 также связаны с нарушением реакции на ЛПС, но отсутствие TLR4 у людей не влияет на исход бактериального сепсиса [14, 15]. К внутриклеточным рецепторам относятся TLR3, 7, 8 и 9. Они распознают различные мотивы нуклеотидных последовательностей и обеспечивают противовирусную защиту. Активация TLR7 и TLR8 приводит к транскрипции интерферонового фактора регуляции – 3 (IRF3) и IRF7 и последующей продукции интерферонов I типа.
TLR3
TLR 3 экспрессируется, в основном, в миелоидных дендритных клетках [16-18]. Хотя TLR3 является внутриклеточным рецептором, показано, что эпителиальные клетки и макрофаги могут также экспрессировать рецептор на своей поверхности [15]. Рецептор распознает различные нуклеотидные последовательности и обеспечивает противовирусную защиту [16, 17].
Установлено, что TLR3 является детектором двухцепочечной РНК [19, 20]. Считается, что TLR3 распознает вторичную структуру РНК, и также вызывает продукцию 1-го типа интерферонов (ИФН) и провоспалительных цитокинов. Двухцепочечная РНК вирусов вызывает созревание дендритных клеток посредством TLR3 [19]. По-видимому, эта РНК может выступать в качестве естественного адъюванта, который содействует потере толерантности против представленных эндогенных или экзогенных антигенов и модулирует баланс Т-клеток-хелперов – Th1/Th2 в последующем их ответе. Среди множества моноцитарных иммунных клеток, TLR3 экспрессируется на мышиных макрофагах, в то время как у человека экспрессия TLR3 осуществляется исключительно на миелоидных дендритных клетках [19, 21, 22]. Кроме того, TLR3, как сообщается, экспрессируется на большом числе неиммунных клеток, включая клетки клубочкового мезангия [23], астроциты [24], клетки маточного эпителия [25] и фибробласты [26].
TLR7 и TLR8
Как и TLR3, TLR7, TLR8 и TLR9 находятся внутриклеточно в эндосомах и распознают фагоцитированные лиганды [27, 28]. TLR7 и TLR8 распознают вирусные РНК, а также различные синтетические аналоги гуанозина [1, 29]. Они активируют дендритные клетки, которые созревают и продуцируют провоспалительные цитокины [29].
TLR9
Неметилированной цитозин-гуанозин (CpG) ДНК является важным лигандом для TLR9 [30]. CpG динуклеотид является стимулирующим мотивом бактериальной и вирусной ДНК [31]. CpG-ДНК является В-митогеном клетки и сильным активатором дендритных клеток в организме человека [28]. В комплексе с другими белками он вызывает усиление антиген-специфического гуморального и Th1-клеточного иммунного ответа [32]. TLR9 находится в эндоплазматической сети и перераспределяет эндосомы при взаимодействии с CpG-ДНК [27]. Производство синтетических CpG-олигодезоксинуклеотидов явдяется мощным инструментом для проведения исследований в этой области. В недавнем исследовании в качестве еще одного природного лиганда для TLR9 был описан плазмодий. Это внесло сомнения в концепцию, согласно которой TLR9 признает специфические последовательности нуклеиновых кислот [33, 34], которая подтверждается тем фактом, что образованные наночастицы ДНК могут модулировать TLR9 сигнализацию на производство высокого уровня 1 типа ИФН [35].
TLR10 и TLR11
TLR10 экспрессируется на различных клетках человека, но его лиганды, а также последствия активации еще предстоит установить [36]. TLR11 был открыт сравнительно недавно [37]. Согласано имеющимся данным, TLR11 распознает молекулы уропатогенных E.coli и профилин-подобные молекулы Toxoplasma gondii [37, 38], однако полный спектр распознаваемых микроорганизмов до конца еще не определен. Опубликованные данные по гену TLR11 включают стоп-кодон в кодирующей последовательности [37].
Экспрессия TLR10 преимущественно осуществляется на В-клетках и плазмацитоидных дендритных клетках миндалин. Он способен формировать гомодимеры и гетеродимеры с TLR1 и TLR6.
Специфика TLR сигнализации
Специфика TLR сигнализации зависит от экспрессии конкретного типа клеток, потенциала для гетеродимеризации определенных TLR и группы цитоплазматических адаптерных молекул [1]. Фактор 88 (MyD88) миелоидной дифференцировки (первичного ответа) был первым идентифицированным адаптером [39]. Исследования с MyD88-дефицитными мышами показали, что существуют другие, MyD88-независимые сигнальные пути, связанные с Toll-IL-1 рецептором (TIR), домен-содержащим адаптером TRIF (TIR домен-содержащий белковый адаптер, вызывающий продукцию бета-интерферона) и TRAM (TRIF связанный адаптер). MyD88 является единственным адаптером для TLR9, но TLR2, TLR4 и TLR6 могут использовать TIR домен-содержащий адаптер или адаптер MyD88. Соответственно, активация TLR3 и TLR4 может включать в себя MyD88 или TRAM [39] (рисунок). Далее в процесс TLR-сигнализации вовлекаются члены семейства IL-1 рецептор-ассоциированных киназ и IFN-регуляторный фактор, которые в конечном счете активируют фактор транскрипции ядерного фактора (NF)-kB и белки из семейства ИФН-регуляторных факторов [39]. Генетические дефекты в механизмах TLR-сигнализации чаще вызывают иммунодефицит, но не аутоиммунный синдром [40]. Однако, недавнее исследование, включающее анализ связей в 44 одиночных нуклеотидных полиморфизмов в 13 генах 1-го типа ИФН определили два полиморфизма тирозинкиназы 2 и IFN-регуляторном факторе 5 генов, которые связаны с системной красной волчанкой [41].
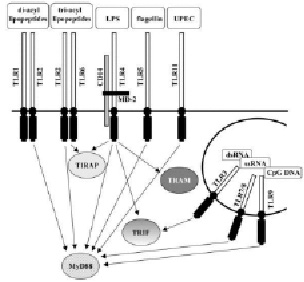
Схема передачи сигналов через TOLL-рецепторы в клетку
Связываясь с PAMPs, TLRs инициируют мощные механизмы врожденного антимикробного иммунитета. Сигнал с TLRs через MyD88 вызывает NF-kB-зависимую продукцию провоспалительных цитокинов и хемокинов, которые вызывают местное и системное воспаление, включая артрит [42, 43]. Например, как CpG ДНК, так и ЛПС могут вызвать активную продукцию фактора некроза опухоли альфа и других провоспалительных медиаторов у мышей, в то время как TLR4-дефицитные и TLR9-дефицитные мыши не реагируют на соответствующие лиганды [21, 30]. Вирусные нуклеиновые кислоты связывают определенный набор TLRs. Например, сигналы от TLR3 через адаптер TRIF, который вызывает продукцию 1-го типа ИФН, одного из основных компонентов противовирусного иммунитета [44]. Сигналы с TLR9 через MyD88 также стимулируют производство большого количества 1-го типа ИФН после распознавания связанных вирусных CpG-ДНК [35]. После связывания с TLRs, антиген-представляющие клетки с участием костимулирующих молекул выделяют провоспалительные цитокины. Связывание TLRs, в основном, вызывает секрецию Th1 цитокинов, которые впоследствии управляют T-клеточной функцией, воздействуя на Th1-тип иммунитета [45-47]. Таким образом, TLRs лиганды выступают в качестве вакцинных адъювантов [48, 49].
Распознавание патогенов паттерн-распознающими рецепторами (ПРР) при СД1
Известно, что хемокины являются центральными медиаторами клеточных перемещений и участвуют в развитии СД1 как у NOD–мышей, так и у человека. Было показано, что мышиные и человеческие клетки островков, включая производящие инсулин ß-клетки, экспрессируют TLR, и данный механизм увеличивает продукцию провоспалительных хемокинов. Эти результаты позволили предположить, что среди инициирующих факторов в развитии СД1 микробные продукты способны активировать TLR островков посредством продукции хемокинов, которые привлекают в очаг воспаления Т-клетки, макрофаги и дендритные клетки. Таким образом, TLR-индуцированная продукция хемокинов может быть важным событием в реализации ранних механизмов развития СД1, приводя к лейкоцитарной инфильтрации островков Лангерганса [50].
Инсулит у человека протекает менее интенсивно по сравнению с инсулитом в животных моделях и морфологически характеризуется, в основном, Т-клетками, хотя В-клетки, макрофаги и NK клетки также присутствуют в изучаемых образцах.
Медиаторы воспаления могут способствовать длительному функциональному подавлению и гибели β-клеток, модуляции регенерации островковых клеток и, как следствие, – их апоптозу. Адаптивная иммунная система распознает весьма разнообразные антигены микроорганизмов посредством поверхностных Т-клеточных рецепторов. В последнее время было замечено, что человеческие панкреатические островки экспрессируют TLR2, TLR3, TLR4 и TLR9 [1].
Во время вирусной инфекции появляется двухцепочечная РНК, которая накапливается в цитоплазме, связывается с TLR3 и выступает в качестве триггера ряда реакций, которые приводят к апоптозу β-клеток через активацию транскрипционных факторов, таких как NFkB и IRF-3. Эти факторы транскрипции увеличивают экспрессию матричной РНК провоспалительных цитокинов, в частности, IFNα, IFNγ, ИЛ-1β, ФНО и хемокинов (например, CXCL10), которые привлекают моноциты, Т-лимфоциты и NK клетки, тем самым увеличивая воспаление в островках и, как следствие, – дисфункцию и повреждение β-клеток [51].
TLR2 иTLR4 соответственно связывают компоненты грам-положительных и грам-отрицательных бактерий. Они находятся в различных клетках и тканях, преимущественно на моноцитах [52].
Основной причиной смерти при СД1 является атеросклероз и воспаление, которое играет ключевую роль в его развитии. При этом гипергликемия способствует развитию сосудистых осложнений диабета, вызывая продукцию цитокинов, хемокинов и активацию NFkB. Сочетание воспаления, гипергликемии и диабета имеет тесную связь с иммунной системой [53]. Количество матричной РНК для TLR2 и TLR4 значительно увеличивается в условиях гипергликемии [54]. Согласно другим исследованиям, экспрессия TLR2 и TLR4 увеличивается на моноцитах пациентов с СД1 [55].
Среди лигандов, которые могут связываться с TLR2 и TLR4, эндотоксин является основным экзогенным лигандом для TLR4. Уровни эндотоксина значительно повышаются у пациентов с СД1 по сравнению с контрольной группой и могут привести к воспалению вследствие активации TLR4 [56]. Кроме того, течение СД1 сопровождается повышением уровня Hsp60 (белок теплового шока 60), который может индуцировать продукцию провоспалительных цитокинов с последующей активацией внутриклеточных сигнальных путей через взаимодействие с тремя распознающими рецепторами: TLR2, TLR4 и RAGE и регуляцию генов, участвующих в провоспалительных реакциях, таких как IL-1, ФНО-α, и MCP-1 в моноцитах [57].
На основании анализа сыворотки крови пациентов с СД1 было показано, что хемокины, продуцируемые в ответ на связывание лиганда с TLR4, участвуют в развитии воспаления, направляя миграцию лейкоцитов и вызывая их активацию на ранних стадиях иммунного реагирования, способствуя переходу к адаптивному иммунитету. В частности, в сыворотке крови пациентов с сахарным диабетом 1 типа, наблюдалось повышение концентрации хемокинов, таких как CCL3, CCL4 и CXCL10 (IP10) [1]. CXCL10 является мощным хемоаттрактантом. Он модулирует экспрессию молекул адгезии после ИФН-γ стимуляции. Связывания ЛПС TLR4 индуцирует продукцию CXCL10 с привлечением NFkB [54]; эти молекулы привлекают больше моноядерных клеток, что приводит к продукции нескольких видов цитокинов. Этот порочный круг может привести к прогрессивному и селективному накоплению макрофагов и Т-клеток в островке, а затем разрушению β-клеток поджелудочной железы.
Роль НК-клеток (связь других факторов врожденного иммунитета с TLRs)
В последние годы появляется все больше данных, свидетельствующих о том, что врожденный и адаптивный иммунный ответ участвуют в патогенезе сахарного диабета 1 типа с привлечением не только Т- и В-лимфоцитов, но и других компонентов врожденной иммунной системы, таких как НК-клетки и дендритные клетки. Как следствие, – модуляция врожденной иммунной системы может рассматриваться в качестве потенциально интересной стратегии лечения и профилактики сахарного диабета 1 типа.
Например, ингибирующее действие IL-1 имеет клиническую эффективность при многих воспалительных заболеваниях, включая ревматоидный артрит и даже диабет 2 типа. IL-1 играет роль в β-клеточной дисфункции и разрушении через NFκB сигнализацию, что приводит к клеточному стрессу и последующему апоптозу β-клеток. Кроме того, ИЛ-1 действует на иммунную систему, влияя на взаимодействие между врожденным и адаптивным иммунным ответом. Соответственно, он представляет собой потенциальную мишень интервенции в аутоиммунный диабет [58].
Большой интерес также представляет модуляция анти-островковой иммунной агрессии. Это касается взаимодействия между микробами и компонентами врожденной иммунной системы. В недавних исследованиях было показано, что NOD мыши, лишенные MyD88 белка, не развивают СД1 [59]. Этот эффект зависит от наличия синантропных микробов, а MyD88 дефицит изменяет состав дистальной кишечной микрофлоры. Предполагается, что взаимодействия кишечных микробов с врожденной иммунной системой могут выступать в качестве критических эпигенетических факторов, влияющих на предрасположенность к СД1. Исследования на животных моделях и у человека показали возможное участие естественных киллеров не только в развитие болезни, но и в защите от нее. Таким образом, предполагается, что эти клетки играют двойную роль в патогенезе СД1. Возможно, это связано с гетерогенностью данного заболевания. Обсуждается также вероятность различного вклада НК клеток в патогенез заболевания в зависимости от стадии болезни.
Заключение
TLRs являются ключевыми рецепторами распознавания микробных, вирусных и грибковых агентов. Их конкретная роль в формировании врожденного и адаптивного иммунитета заключается в механизмах поддержания толерантности в организме хозяина. Связывание TLRsможет способствовать потере толерантности по нескольким механизмам. Конкретные роли отдельных сигнальных путей для различных аутоиммунных состояний еще предстоит установить. Предварительные исследования с функциональными антагонистами TLRsпоказали, что TLRsмогут выступать в качестве набора потенциальных мишеней для лечения и профилактики многих аутоиммунных заболеваний и их осложнений и, в частности, СД1.
Исследования молекулярных механизмов участия NK клеток в регуляции аутоиммунных процессов в островках Лангерганса необходимо продолжать и углублять с учетом ранее полученных противоречивых результатов. Это необходимо для разработки новых терапевтических стратегий, направленных на профилактику и/или лечение СД1.
Библиографическая ссылка
Репина Е.А. ФАКТОРЫ ВРОЖДЕННОГО ИММУНИТЕТА ПРИ САХАРНОМ ДИАБЕТЕ 1 ТИПА // Международный журнал прикладных и фундаментальных исследований. 2016. № 5-1. С. 72-79;URL: https://applied-research.ru/ru/article/view?id=9185 (дата обращения: 02.08.2026).